




- @C20180602_csq
简介
该用户还未填写简介
擅长的技术栈
可提供的服务
暂无可提供的服务
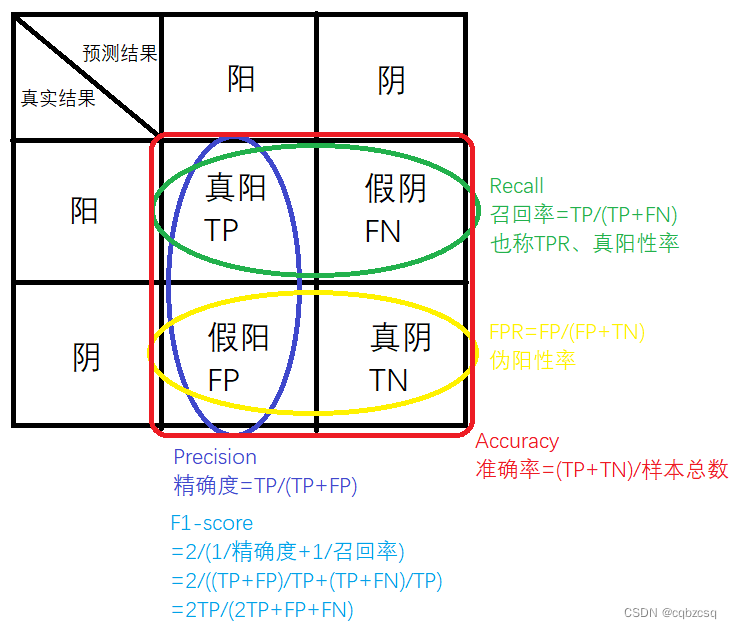
TP、FP、TN、FN准确率、精确率(查准率)、召回率(查全率)真阳性率TPR、伪阳性率FPRF1-score=2TP/(2*TP+FP+FN)最大响应分数Fmax为F1-score的最大值(在最佳阈值的前提下)
本文记录了使用Salmon、tximport和DESeq2进行转录组分析的完整流程。首先安装Salmon并下载参考基因组数据,建立索引后分析测序数据,输出TPM等表达量指标。接着构建R语言环境,安装tximport和DESeq2等工具,处理GTF文件建立基因映射关系。然后通过tximport构建基因表达矩阵,使用DESeq2进行差异表达分析,比较实验组与对照组的基因表达变化。最后介绍了WGCNA共
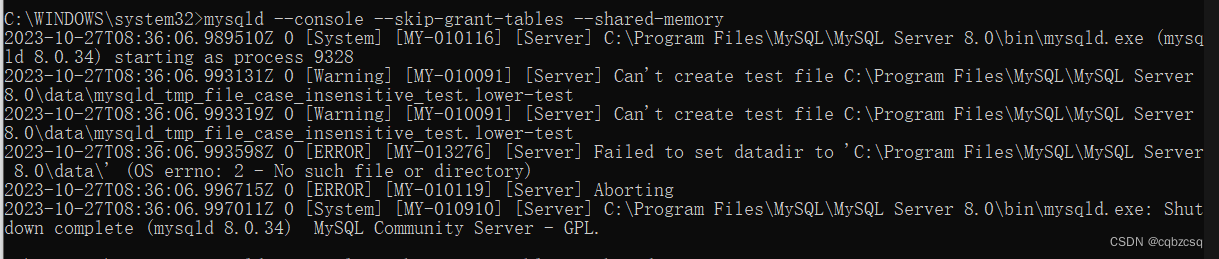
我的MySQL启动可以直接连接,但是就是无法使用密码验证,后来出现pymysql和MySQL连接的时候出现了无法鉴权的问题:pymysql.err.OperationalError: (1045, "Access denied for user 'root'@'localhost' (using password: YES)")后来发现是mysqld无法直接运行,原因应该是安装的时候发生了一些问题
每次使用vscode写python代码的时候,都需要在外面跑一个anaconda prompt,激活环境,然后进入对应的文件夹,运行代码,特别麻烦,所以想,能不能直接在vscode终端里面激活环境然后运行。

之前介绍了ESMC-6B模型的网络接口调用方法,但申请token比较慢,有网友问能不能出一个本地部署ESMC小模型的攻略,遂有本文。其实本地部署并不复杂,官方github上面也比较清楚了。后面讲述了esmc-decode接口的使用方式。

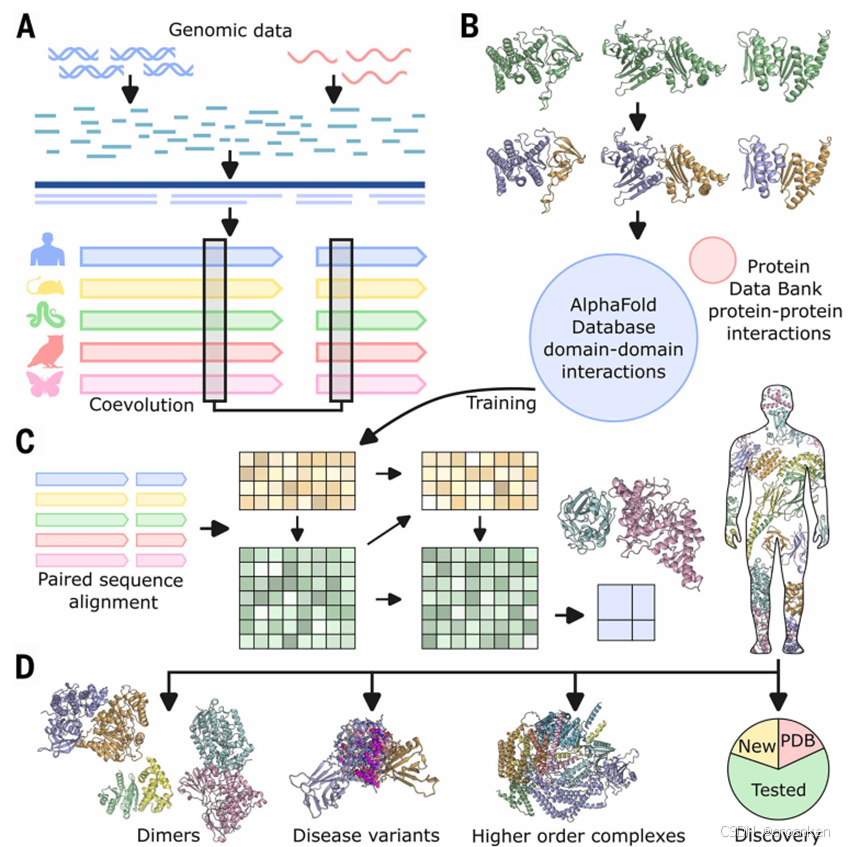
本文介绍了一种预测人类蛋白质组中蛋白质相互作用(PPI)的创新方法。研究团队开发了RF2-PPI深度学习模型,结合omicMSA共进化分析和AlphaFold2结构预测,实现了大规模PPI筛选。该方法首先构建了包含2万多个物种的蛋白质组数据库,通过改进的pMSA算法分析蛋白质共进化关系;然后从AlphaFold数据库挖掘结构域间相互作用(DDI)数据;最后利用简化的RoseTTAFold2架构预测
每次使用vscode写python代码的时候,都需要在外面跑一个anaconda prompt,激活环境,然后进入对应的文件夹,运行代码,特别麻烦,所以想,能不能直接在vscode终端里面激活环境然后运行。
之前介绍了ESMC-6B模型的网络接口调用方法,但申请token比较慢,有网友问能不能出一个本地部署ESMC小模型的攻略,遂有本文。其实本地部署并不复杂,官方github上面也比较清楚了。后面讲述了esmc-decode接口的使用方式。
PDB文件设计得非常好,能够比较完整地记录实验测定数据从蛋白质结构来看,首先它会有多种不同的测定模型,然后每个模型中包含多条链,每条连上包含若干个残基,每个残基包含若干个原子在biopython.PDB包中可以找到这些概念对应的模块:model、chain、residue、atom首先用PDBParser读取文件,获得structurestruct内部的一层结构是model,我们只取第一个mode

本文主要总结了机器学习课程中的大部分知识点,包含概念学习、决策树、ANN、贝叶斯推理、无监督学习、基于实例的学习、回归学习、线性分类器、特征选择与稀疏学学习。包含有LMS算法、Find-S算法、候选消除算法、ID3、C4.5、朴素贝叶斯、Gibbs算法、Kmeans、层次聚类、KNN、径向基函数RBF、线性回归、逻辑回归、softmax回归、SVM、Widrow-Hoff算法、relief-F算法
