- @weixin_49214410
简介
该用户还未填写简介
擅长的技术栈
可提供的服务
暂无可提供的服务
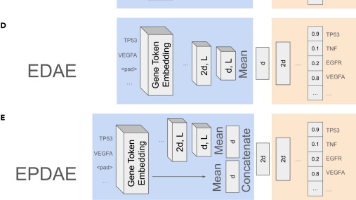
热门的领域都有相应的数据量积累了,比如说基因组、单细胞空转啥的,像 AlphaGenome/evo2/scoby/Borzoi 就是经典的基因组模型,而 Geneformer/scGPT/CellFM 啥的则是经典的单细胞基础模型。今天看到一个今年5月21日刚刚见刊 Cell 子刊 Patterns 的基础模型 GSFM,模型侧重的角度还是比较新颖的,叫做基因集基础模型。补充一下,基础模型最强悍的
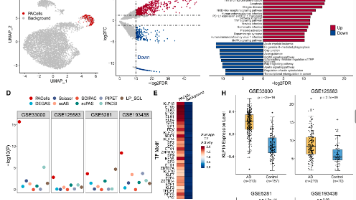
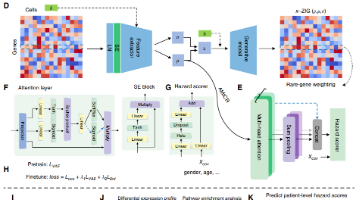
单细胞层面,能够更加明确的看到肿瘤微环境中的异质性细胞群体,数据层面也扩展到一个样本对应数万个细胞。逻辑上,患者的预后不是由所有细胞平均决定的,而是由某些关键细胞亚群决定的。对应的数据格式是很清晰的,每个样本的基因表达以及生存事件,在此基础上可以用 Cox 模型预测患者级别的预后风险。图:scSurvival 模型框架 (A-H) 与具体下游的应用(I-K),其实模型还是相对简单的,并且框架还是比
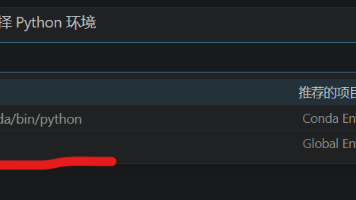
Vscode 连接远程服务器 ipynb 选择内核无法自动显示 conda 环境对应的 python可以看到目前只有base,没有其它conda环境的python解释器选项。
热门的领域都有相应的数据量积累了,比如说基因组、单细胞空转啥的,像 AlphaGenome/evo2/scoby/Borzoi 就是经典的基因组模型,而 Geneformer/scGPT/CellFM 啥的则是经典的单细胞基础模型。今天看到一个今年5月21日刚刚见刊 Cell 子刊 Patterns 的基础模型 GSFM,模型侧重的角度还是比较新颖的,叫做基因集基础模型。补充一下,基础模型最强悍的
从二分类建模、评估、SHAP可解释性甚至到。

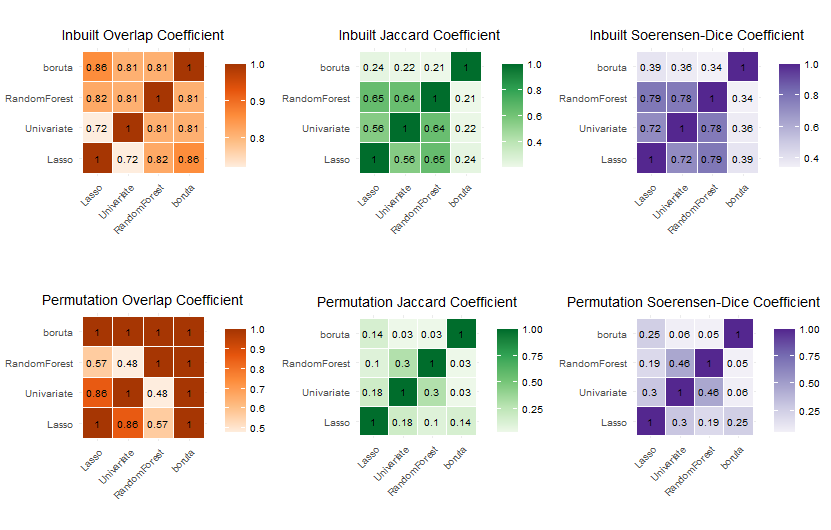
GeneSelectR 包的特征选择过程是使用 Python 的 scikit-learn 库实现的。该软件包需要安装 Anaconda 才能正常工作首次启动时,需要为包创建conda工作环境每次分析需要通过设置正确的 conda 工作环境来重新启动 GeneSelectR 分析该包支持其它来自sklearn库的算法。举个例子,如果您想使用 XGBoost 分类器而不是默认的随机森林# 执行分析y
STAMP方法通过将复杂的基因干扰预测问题分解为三个子任务,展示了与人类认知过程相似的“分而治之”策略。这种方法灵活的借助了当前单细胞扰动领域主流的深度学习模型(如图神经网络、Transformer等),同时其在解决高维数据问题上的思路有相通之处。相较于传统的单一任务模型,STAMP的子任务设计使得其更适应于处理数据维度高且具有稀疏性特征的问题。此外,STAMP与现有的基因调控网络建模方法(如Ce
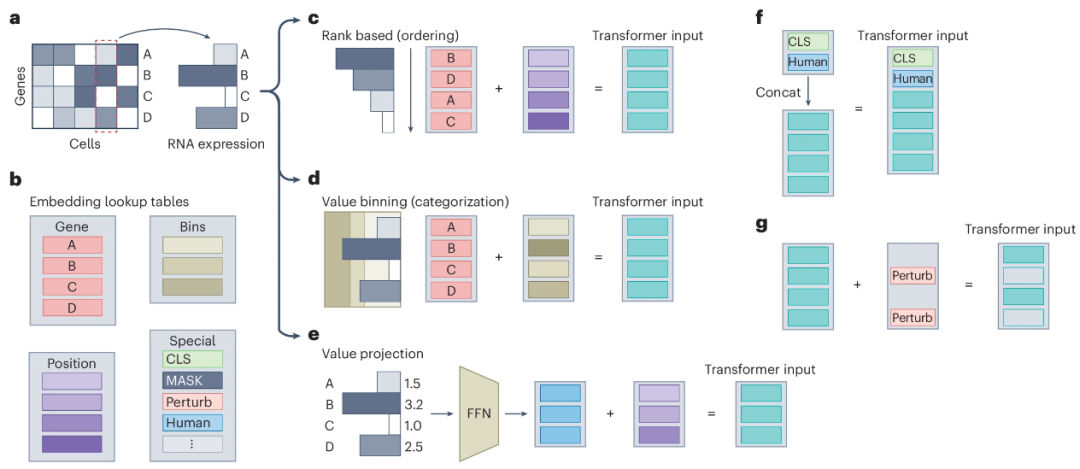
GeneGPT的核心思想是通过大语言模型(如GPT-3.5)对基因信息进行嵌入,从而生成能够反映基因功能和细胞特征的嵌入向量(其他方法则是需要使用大规模的单细胞数据进行预训练,GeneGPT 属于是触类旁通、借花献佛了)。这些基因嵌入向量能够帮助我们在单细胞转录组学研究中更好地表示基因和细胞的生物学信息,被进一步用于细胞级别的嵌入表示。具体来说,基因嵌入通过提取NCBI基因数据库中的文本摘要,并利
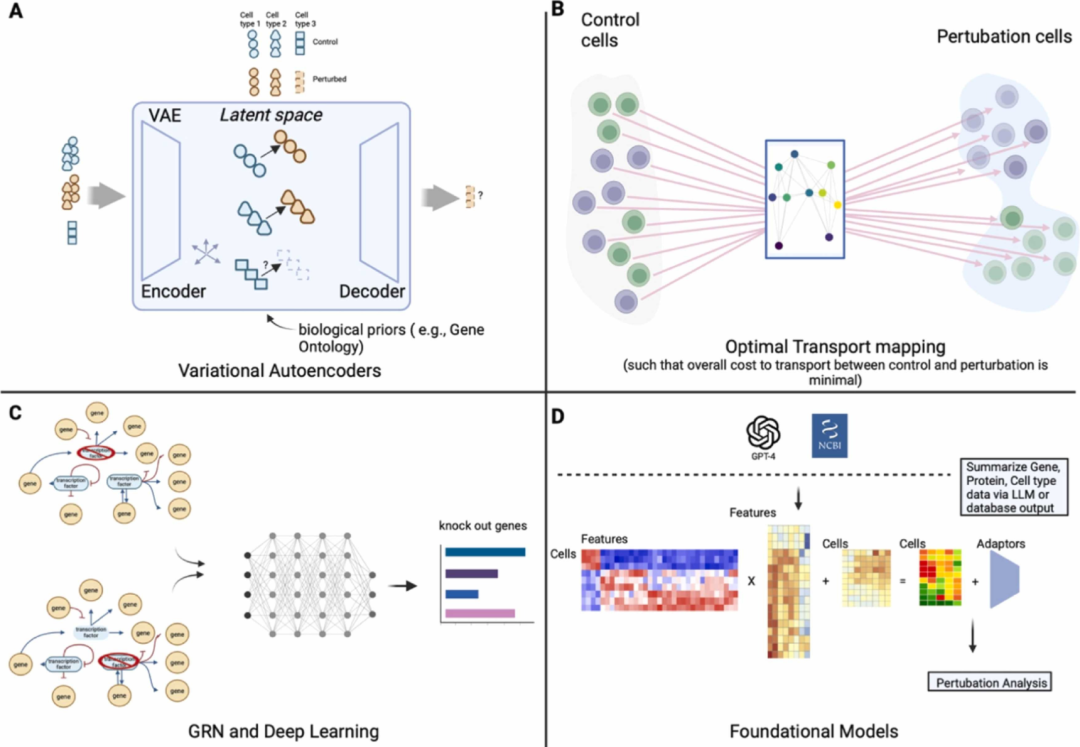
如果对细胞进行一个或多个基因敲除扰动(红色部分,比如通过实验干预特定基因的活性),会导致一些基因表达水平发生变化(紫色部分),这种变化可能是增强、减弱或保持不变。先进的深度学习方法,如基于 transformer 的基础模型,被认为能够学习单细胞中的基因表征,这些表征可以用于预测未见实验的结果,例如预测基因扰动敲除对其它基因转录表达的影响。与之相对, "加性模型" 则采用传统的线性思维,认为组合扰
最近的一个热点那肯定是 bulk 与单细胞的联合分析,不过肯定不是差异+单细胞注释分群,而是直接意义上的通过算法建模两种数据。除了 ATAC 数据以外,作者也在黑色素瘤的单细胞转录组的数据中进行了示例分析,可以看到与其它方法相比更加联系的疾病关联细胞鉴定。在此基础上,根据下面的流程构建 bulk 样本与每个单细胞之间的相似性矩阵,再衡量每个细胞与目标表型之间的关联强度,最后将候选细胞与表型一起构建