- @H20230717
简介
该用户还未填写简介
擅长的技术栈
可提供的服务
暂无可提供的服务
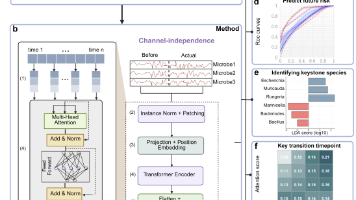
利用稀疏的纵向数据准确预测微生物群落的动态行为,对于基于微生物组的精准医疗和生态监测而言,仍是一项极具挑战性的任务。现有的大多数模型依赖数据插值,并假定是种群层面的动态变化,这限制了它们在现实场景中捕捉个体微生物变化的能力。我们提出了 MicroProphet,这是一种个性化的时序感知框架,能够从不完整的纵向观测数据中准确预测微生物丰度轨迹,且无需数据插补。它由一个时间感知的 Transforme
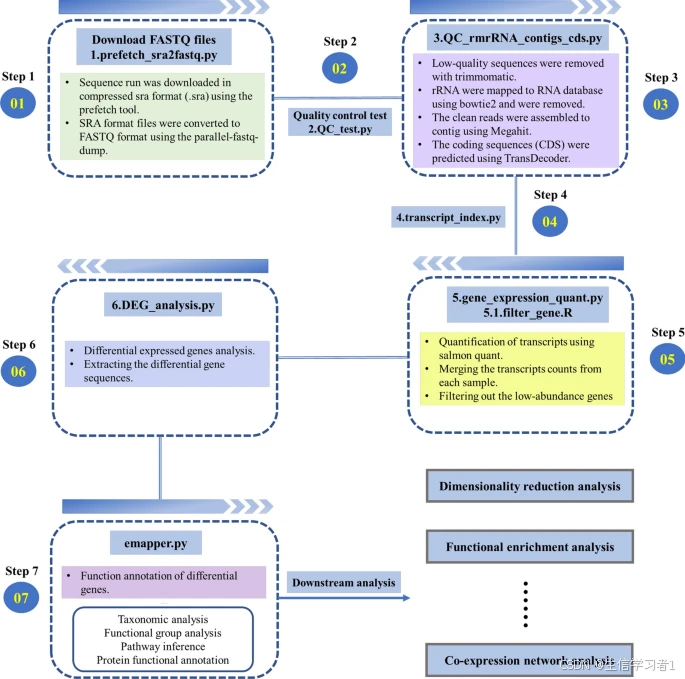
【工具】metaTP:一种集成了自动化工作流程的元转录组数据分析工具包
广义估计方程的基本介绍

【R包】pathlinkR转录组数据分析和可视化利器

转录组数据多来源的批次效应校正方法总结

本文介绍了如何使用R语言实现随机森林算法进行分类任务。随机森林通过构建多棵决策树并投票决定最终标签,具有较高的准确性和鲁棒性。文章详细描述了从数据下载、加载R包、数据切割、调参、建模到模型评估的完整流程。具体步骤包括:1) 下载并导入数据;2) 数据标准化;3) 将数据集划分为训练集和测试集;4) 构建随机森林模型并评估袋外误差(OOB rate);5) 获取特征重要性得分;6) 通过多次建模选择

本文详细探讨了转录组数据分析中常用的差异分析R包(如DESeq2、limma和edgeR)及其与t-test/wilcox-rank-sum test的结合使用。文章首先介绍了如何下载和导入测试数据,并批量安装所需的R包。接着,讨论了基因表达count矩阵的标准化方法(如FPKM、TPM等),以及如何通过PCA、tSNE、UMAP和热图等方法进行基因整体水平分布的可视化。随后,文章分别展示了DES

纵向数据统计方法,广义估计方程和混合线性模型

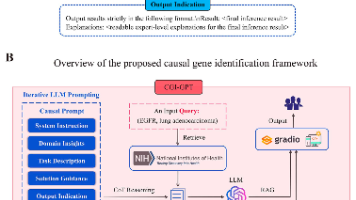
从多组学角度确定与癌症直接相关的基因对于理解癌症的发病机制以及改进治疗策略至关重要。传统的基于广义相关性的统计和机器学习方法用于识别癌症基因,但这些方法往往会产生冗余、有偏差的预测,且解释性较差,这主要是由于忽略了混杂因素、选择偏差以及神经网络中的非线性激活函数所致。在本研究中,我们引入了一种用于在多个组学领域识别癌症基因的新框架,名为 ICGI(整合因果基因识别),它利用一个大型语言模型(LLM
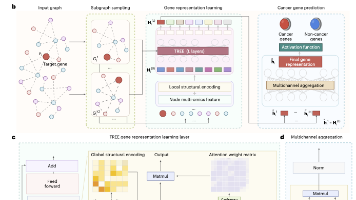
图表示学习已被用于从生物网络中识别癌症基因。然而,其适用性受到解释性和泛化性不足的限制,尤其是在整合网络分析的情况下。在此,我们报告了一种可解释且可泛化的基于转换器的模型的开发,该模型通过利用图表示学习以及将多组学数据与同质和异质生物相互作用网络的拓扑结构相结合,能够准确预测癌症基因。该模型能够解释多组学和更高阶结构特征的各自重要性,其在跨生物网络(包括 miRNA 与蛋白质之间的网络、转录因子与