
【干货指南】几万个Peak不可能都验证,怎么筛出最值得研究的那几个?
做完ATAC-seq、ChIP-seq、CUT&Tag或DAP-seq后,报告里常常会出现成千上万个peak。一开始看结果会很兴奋,这里有peak,那里也有peak,启动子有peak,远端区域也有peak,差异peak 一大堆,motif 结果也列出了一串候选转录因子。但真正写文章、补机制、设计验证实验时这么多peak,不可能每一个都做ChIP-qPCR、双荧光、EMSA或功能验证。那问题就来了。验证资源就那么多,一次ChIP-qPCR能跑的位点有限,下游想接的功能实验更是烧钱烧时间。怎么从这几万个里头,挑出真正值得你押注的那十来个?peak出来之后该用什么逻辑把它们一层层筛下去。
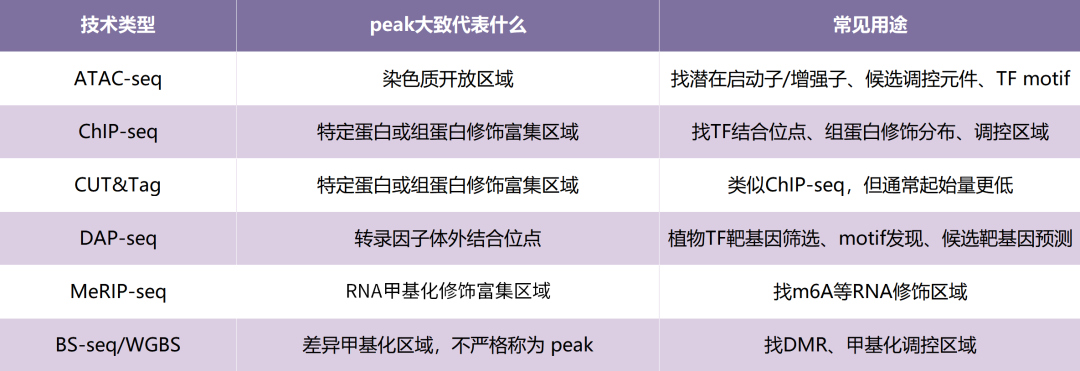
一、不同技术里的peak代表什么含义
不同技术的peak,能说明的问题不同。
二、别急着挑基因,先确认数据质量是否可以用
这一步最容易被跳过,但它决定了后面所有工作是不是白做。在动peak之前,先把整体数据质量过一遍,确认信号是真的。
(1)测序质量和比对率。这是地基,比对率太低、重复率异常高的数据,后面的peak本身就不可信。
(2)样本间相关性。 重复样本之间相关性一般都不错,如果两个生物学重复差异很大,说明哪个环节可能有问题,先别急着分析数据。
(3)peak在基因组功能区的分布。 重点看promoter-TSS区域有没有合理占比。转录因子和很多组蛋白修饰的核心调控功能就发生在TSS附近,如果你做的是TF,promoter区却几乎没有富集,那要警惕抗体特异性或者结合模式的问题。
(4)peak在TSS上下游的分布。 把IP的reads信号沿TSS上下游画出来,正常情况下应该在TSS附近形成明显富集峰。这条曲线漂不漂亮,基本能一眼判断这批数据值不值得深挖。
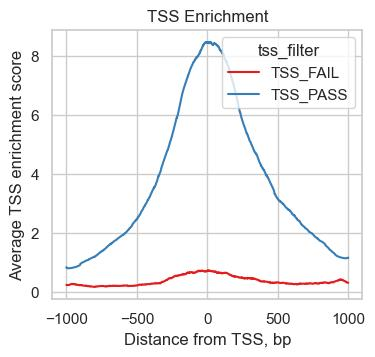
图 TSS附近reads富集情况示意图。正常情况下,优质ChIP-seq/ATAC-seq数据应在TSS附近形成明显富集峰。
(5)FRiP 值。 落在peak内的reads占总reads的比例,反映信噪比。经验上5%以上算可以接受,这条不达标,假阳性会非常多。
(6)peak 数目是否符合预期。 转录因子通常会有上千个结合位点(经验上T 大于1000个peak 比较正常),组蛋白修饰则看修饰类型。数目过少要怀疑富集失败,过多要怀疑背景太高。
(7)重叠peak占比。 重复样本之间,重叠peak占比50%以上,说明结果的可重复性是过关的。
这一步走完,你心里应该有底了:这批数据能不能信,值不值得往下挑。
三、motif分析——这是TF数据最需要关注的一张表
如果你做的是转录因子,这一步的重要性怎么强调都不过分。它能直接告诉你:你拉下来的,到底是不是你想要的那个蛋白。打开HOMER的Known Motif富集结果,重点看这几部分内容:
(1)排在最前面的motif是不是你的目的蛋白,或者同一家族。 软件会把富集到的known motif按显著性(P-value)从高到低排好。如果排名第一的motif匹配到的正是你研究的TF,或者它所属的转录因子家族,那这批数据的可信度直接拉满。如果完全对不上那就要继续关注一下其他质控指标。
(2)一般第一个motif 要明显比第二个更显著。 一个干净、特异的 ChIP 数据,头号motif的显著程度应该和后面的拉开差距。如果前几个motif显著性差不多、谁也不突出,可能结合特异性可能不够。
(3)避开假motif。 碱基复杂度低、只有两个碱基反复重复那种(比如 GAGAGA、CTCTCT),通常不是真正有意义的结合基序,不建议优先考虑。优先挑序列复杂、信息量高的motif。
motif 这一关过了,你才有底气说后面挑出来的peak是真的。
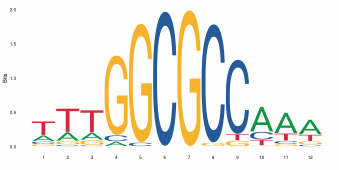
图 JASPAR数据库中转录因子结合Motif示意图。
四、开始真正缩圈——把几万压到几十
确认数据可信之后,正式收紧范围。这一步有六个步骤参考,可以叠加使用,每叠一层,范围就小一圈:
(1)重复样本取交集。 只保留在所有生物学重复里都稳定出现的peak。靠运气出现一次的噪音,在这一步就被砍掉了。这是性价比最高的一刀。
(2)顺着最可信的motif反查peak。 在motif分析里锁定了那个最可信的motif之后,把包含这个motif的peak单独提出来做后续分析。带着明确结合基序的peak,本身就是质量最硬的一批。
(3)只看promoter区。 把分析范围收到启动子区域。落在promoter的结合位点,最有可能直接调控下游基因转录,功能解释最清晰,验证实验也最好设计。这是最容易出故事的一类。
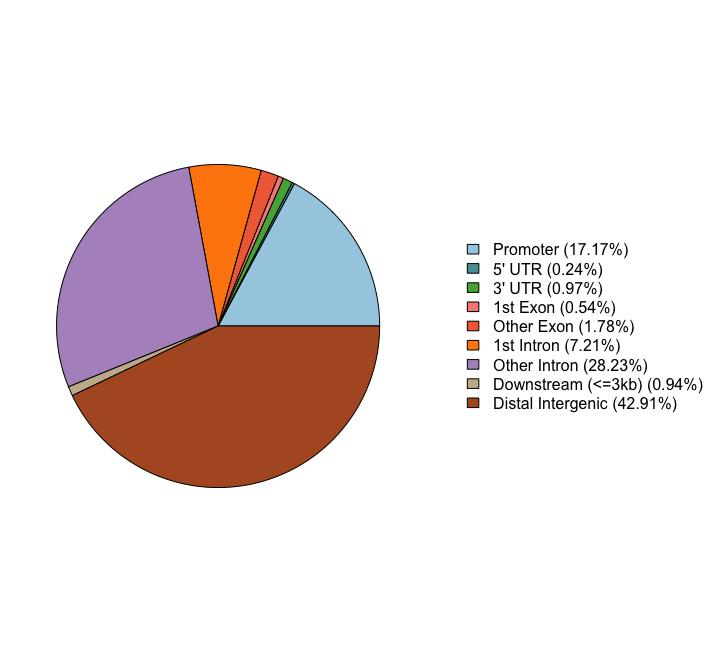
图 Peak在基因组不同功能区域的分布示意图。
(4)提高阈值,只留信号强的peak。 这一步直接卡数值,几个核心列:
——fold enrichment(富集倍数): 这是最关键的排序指标。富集倍数越高,结合越真实可信。
——-log10(p-value): 反映统计显著性。
——-log10(q-value): q值是对p值做多重检验校正后的结果,更严格。
——pileup / abs_summit: pileup反映该位点堆叠的reads数,abs_summit是信号最高点的位置。
把这几列一卡,几千个peak通常能压到几十个又强又显著的核心位点。
(5)做GO / KEGG,锁定关键调控。 把筛出来的peak关联的靶基因拿去做功能富集。看看显著命中的通路里,有没有正好是你课题关心的方向(比如逆境响应、激素信号、发育调控)。命中你关心通路的那些靶基因,就是你的重点验证对象。这一步等于让数据自己告诉你哪些基因最值得做。
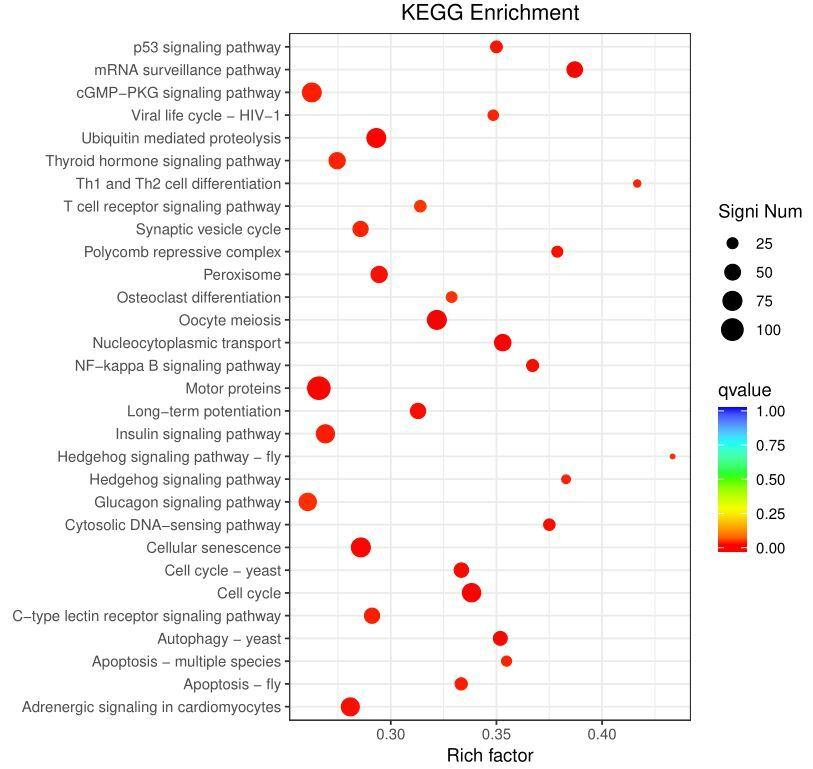
图 GO富集分析示意图,通过功能富集快速定位与研究主题相关的关键通路。
(6)与转录组联合分析(下一节单独讲)。
五、和转录组联合——给你的候选位点上双保险
这是临门一脚,也是审稿人最认的一招。ChIP-seq等技术有个天然的局限:它鉴定的是TF在体外/体内结合了哪些DNA区域,但结合不等于有功能。有些结合可能发生在非活性染色质区,有些可能被其他因子抑制而压根没启动转录。转录组(RNA-seq)正好补上这一块:它告诉你这些基因的表达水平到底变没变。大致逻辑是:
(1)ChIP-seq给你转录因子的直接结合位点 → 潜在靶基因
(2)RNA-seq给你差异表达基因(DEGs)
(3)两个清单取交集
如果一个基因,既被TF结合(ChIP有peak),它的表达又确实随处理发生了显著变化(RNA-seq 是DEG),那就基本能确认这个TF的结合是有真实功能意义的。
取完交集,关键调控基因的范围一下就锁定了。排在最顶上、证据最齐的那些,就是你最该投入实验去验证的对象。
六、挑出最终候选,直接对接实验
筛到这里,假设你已经有了一两个非常感兴趣的基因。最后一步是把它和具体的motif序列对应上,为下游实验做准备。
不同peak对应的验证方式不一样。
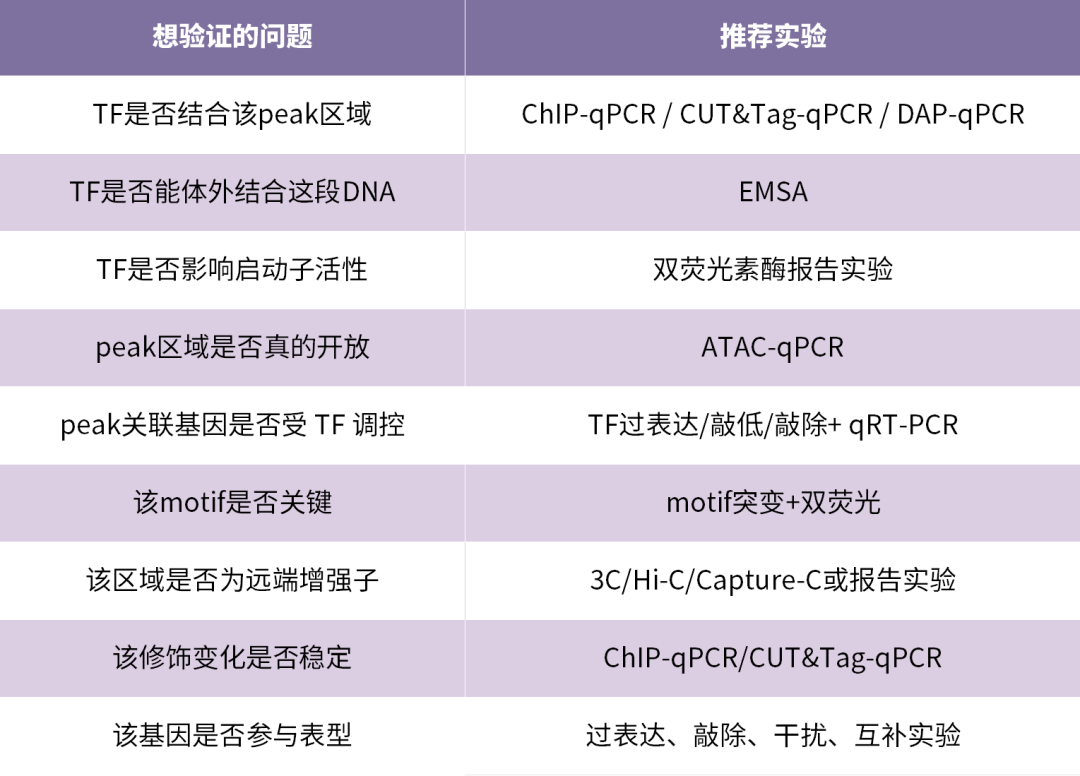
如果经费有限,可以先验证2–5个最高优先级 peak,不建议一开始铺太多。比较常见的验证组合是:
组合 1:ATAC-seq + RNA-seq后筛关键peak
差异开放peak
↓
关联差异表达基因
↓
motif找候选TF
↓
ChIP-qPCR/双荧光验证
↓
构建TF—开放区域—靶基因调控模型
组合 2:DAP-seq + RNA-seq后筛靶基因
DAP-seq peak
↓
启动子区域靶基因注释
↓
与RNA-seq差异基因取交集
↓
筛关键通路基因
↓
DAP-qPCR/EMSA/双荧光验证
↓
构建TF—靶基因—表型模型
组合 3:H3K27ac CUT&Tag/ChIP-seq + RNA-seq后筛增强子
差异H3K27ac peak
↓
关联差异表达基因
↓
结合ATAC-seq判断开放性
↓
筛候选增强子区域
↓
报告实验或ChIP-qPCR验证
↓
解释关键基因表达激活机制
更多推荐
 10
10 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)