- @qq_42491125
简介
该用户还未填写简介
擅长的技术栈
可提供的服务
暂无可提供的服务
文章目录来,我要诱惑你Linux系统与shell安装属于你的Linux系统shell的功能shell特点shell版本shell初体验Hello Shellshell版俄罗斯方块怎么学shell参考作者:余涛email:yutao@big.ac.cn中国科学院大学来,我要诱惑你想要入门生信吗?想要学习Linux系统吗?想要花最少的时间学会一门编程语言?想要写一个shell版俄罗斯方...
文章目录哪些是序列序列常用操作1.取值/索引2.修改3.增加4.删除5.排序6.统计哪些是序列Python的数据类型除了数值(数字)和bool值之外,其他的都是序列对象,或者叫做容器,包括:字符串、列表、元组、集合和字典。它们的共同特点是能够存储一串、一系列的数据。序列常用操作通过序列对象 变量名.方法实现列表不同的操作这里以列表为例,首先声明:boys = [] #使用[]定义一个空...
来自B站《猴博士爱讲课》,摘取行列式6种常用的运算性质1.对角线x,其余a[xa⋯aax⋯a⋮⋮⋱⋮aa⋯x]=(x−a)n−1[x+(n−1)a]\begin{bmatrix}x&a&\cdots&a\\a&x&\cdots&a\\\vdots&\vdots&\dd..
文章目录参考prodigal -i my.metagenome.fna -o my.genes -a my.proteins.faa -p metanohup time prodigal -a HTR8.faa -d HTR8.fna -f gff -o HTR8.gff -p meta -i ../HTR8_Megahit.fa &>HTR8.log &-a:输出选中文件的
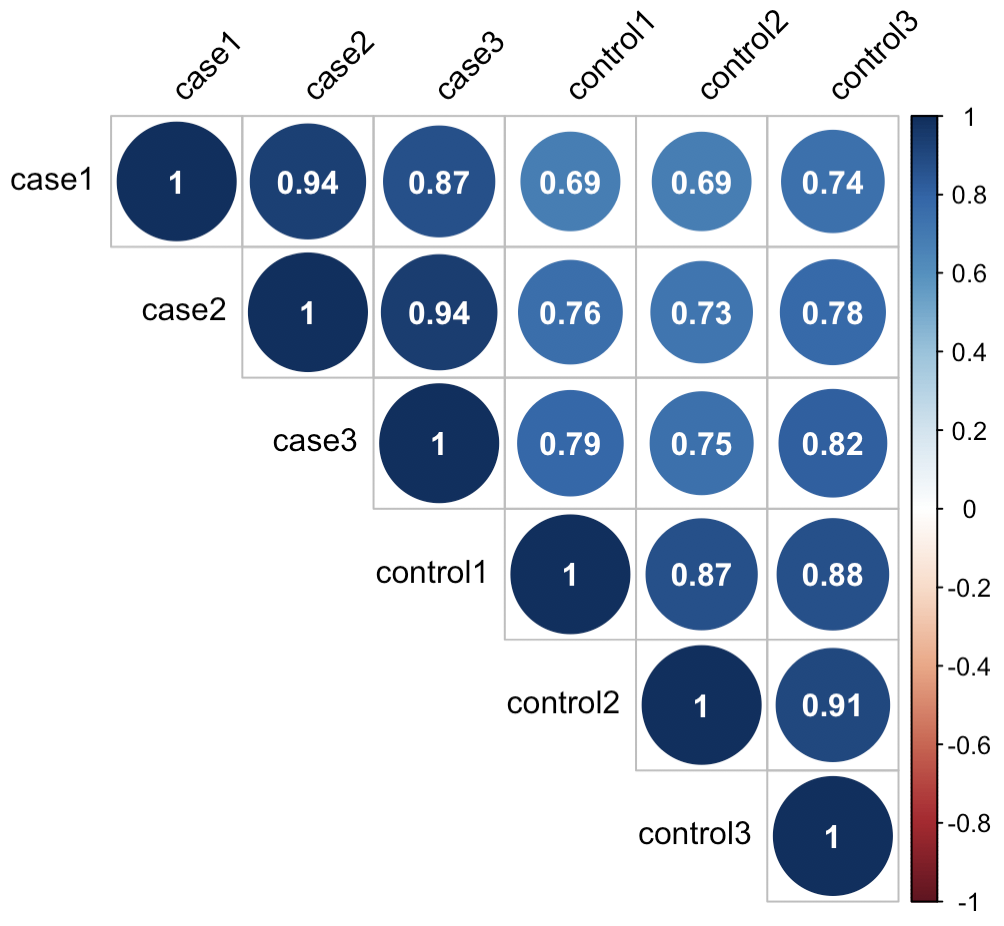
文章目录引言数据计算相关系数映射相关系数到热图完整代码引言生物学实验中,常常需要设置重复,例如技术重复、生物学重复,以此确保不是个体的偶然变异对结果产生影响。以转录组数据为例,一般会设置3-5个生物学重复,如何确认生物学重复的效果好坏呢,方法有很多,可以计算两两样本之间的相关性,可以进行样本的PCA分析,或者绘制聚类热图,这里首先介绍样本相关性方法。我们将在R,使用Rstudio进行计算绘图。数据
文章目录安装使用简介摘要背景介绍安装(gtdbtk) [yutao@myosin Genome_integration]$ mamba create -n orthofinder -c bioconda orthofinder使用简介OrthoFinder: phylogenetic orthology inference for comparative genomicsGenome Biolog
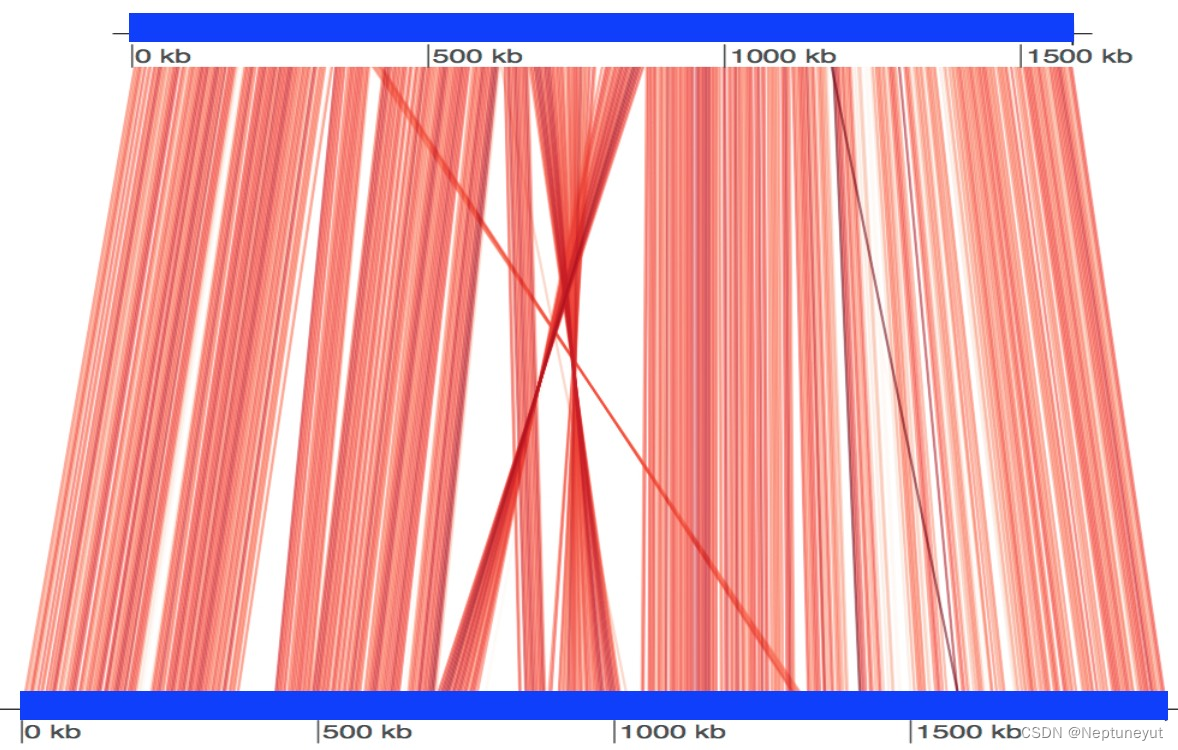
错误信息std::bad_alloc/Exit code -6排错从中断点重新跑:nohup megahit --continue -o Cluster3&>Log/cluster3_coassembly_continue.log &-o 为之前的输出结果目录
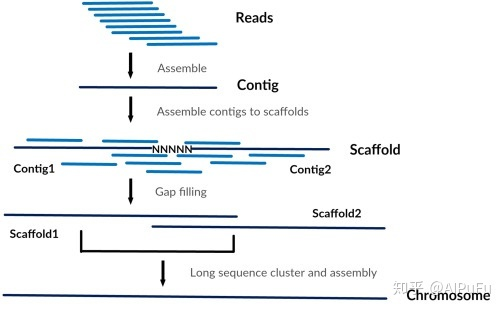
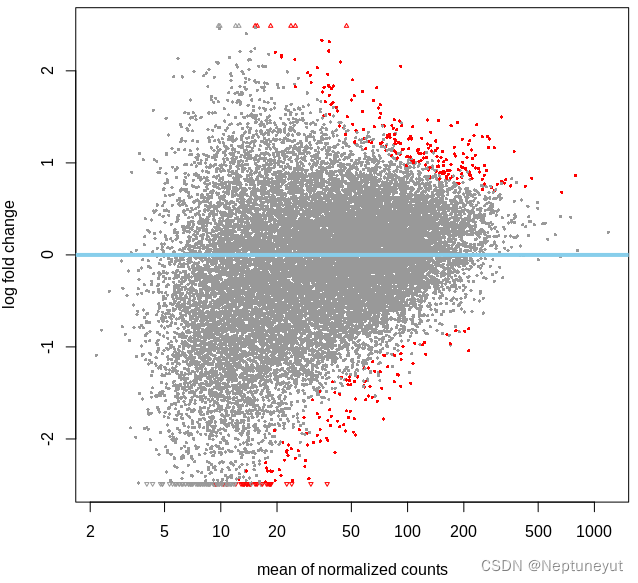
文章目录引言安装并导入DESeq2包数据要求制作dds对象,进行差异分析筛选差异基因完整代码引言对于组学分析来说,常常会寻找组间的差异,例如差异基因(转录组)、差异菌(宏基因组)以及差异通路(宏基因组),而转录组分析上最为经典的DESeq2包对于以上分析也都适用DESeq最早在2010年发表在Genome Biology上,2014年上更新版本DESeq2。DESeq2是基于负二项广义线性模型估算
文章目录为什么用Django不得不说的网页框架模型MVCMTVDjango流程Django项目结构步骤参考为什么用Django如果你目前面对以下情况:时间紧、任务重,要求快速开发一个比较全面的网站不会java,只会python了解一些前端html/css/javascript知识,但是从未真正意义实现过一个网站那么恭喜你,Django为你量身打造Django将功能进行封装,简化调用框架划分成模块,
(gtdbtk) [yutao@myosin Eisenbacteria]$ head fa.idGCA_001780165.1_genomic.faGCA_003235575.1_genomic.faGCA_005893165.1_genomic.faGCA_005893185.1_genomic.faGCA_005893225.1_genomic.faGCA_005893265.1_genom