




- @qq_36608036
简介
该用户还未填写简介
擅长的技术栈
可提供的服务
暂无可提供的服务
一、生成数据表1、首先导入pandas库,一般都会用到numpy库,所以我们先导入备用:import numpy as npimport pandas as pd2、导入CSV或者xlsx文件:df = pd.DataFrame(pd.read_csv(‘name.csv’,header=1))df = pd.DataFrame(pd.read_excel(‘name.xlsx’))3、用pand
fdr padjust 统计计算
植物基因组数据库0.0722018.08.24 21:39:15字数 748阅读 1054| Database | URL | Species || RARTF | http://rarge.gsc.riken.jp/rartf/ | 拟南芥 || AGRIS, AtTFDB | http://arabidopsis.med.ohio-state.edu/AtTFDB/ | 拟南芥 ...
本文主要简要的比较了常用的boosting算法的一些区别,从AdaBoost到LightGBM,包括AdaBoost,GBDT,XGBoost,LightGBM四个模型的简单介绍,一步一步从原理到优化对比。AdaBoost原理原始的AdaBoost算法是在算法开始的时候,为每一个样本赋上一个权重值,初始的时候,大家都是一样重要的。在每一步训练中得到的模型,会使得数据点的估计有对有错,我们就在每一步
ascp 高速下载NCBI各种数据库中的数据(SRA NR NT 分类数据库)NR NT 数据库:#wget -c https://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz#wget -c https://ftp.ncbi.nlm.nih.gov/genbank/livelists/gi2acc_mapping/gi2acc_lmdb.db.gz#w
video标签设置autoplay(自动播放)无效问题: 在使用video标签的时候,给它设置了autoplay属性,发现没有什么效果;解决方式: 给video标签加上muted(静音)属性就可以自动播放了;
高通量测序的方式主要有:单端测序、paired-end/mate-paired(PE/MP)测序高通量测序的方式主要有:单端测序、paired-end/mate-paired(PE/MP)测序 [8] 。当要进行多 个样品同时测序时可以给不同的样品添加不同接头,混合后一起测序。其中单端测序就是将 基因组随机打断后,对每个片段的进行测序。该方式建库简单,操作步骤少,常用于小基因...
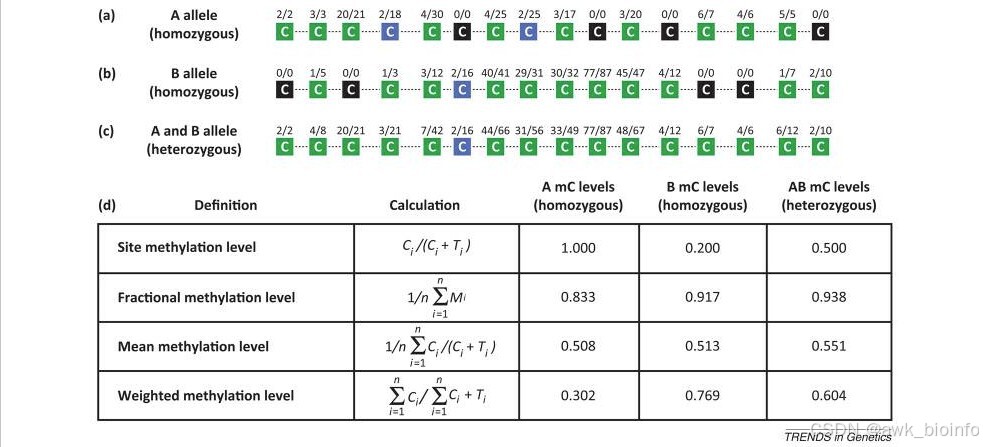
甲基化水平计算
ascp 高速下载NCBI各种数据库中的数据(SRA NR NT 分类数据库)NR NT 数据库:#wget -c https://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz#wget -c https://ftp.ncbi.nlm.nih.gov/genbank/livelists/gi2acc_mapping/gi2acc_lmdb.db.gz#w
先上下面这段代码:const parent = this.el.parentElementconsole.log(parent.children)parent.children.forEach(child => {console.log(child)})会报错:Uncaught TypeError: parent.children.forEach is not a function解决办法一