- @qq_42090739
简介
该用户还未填写简介
擅长的技术栈
可提供的服务
暂无可提供的服务
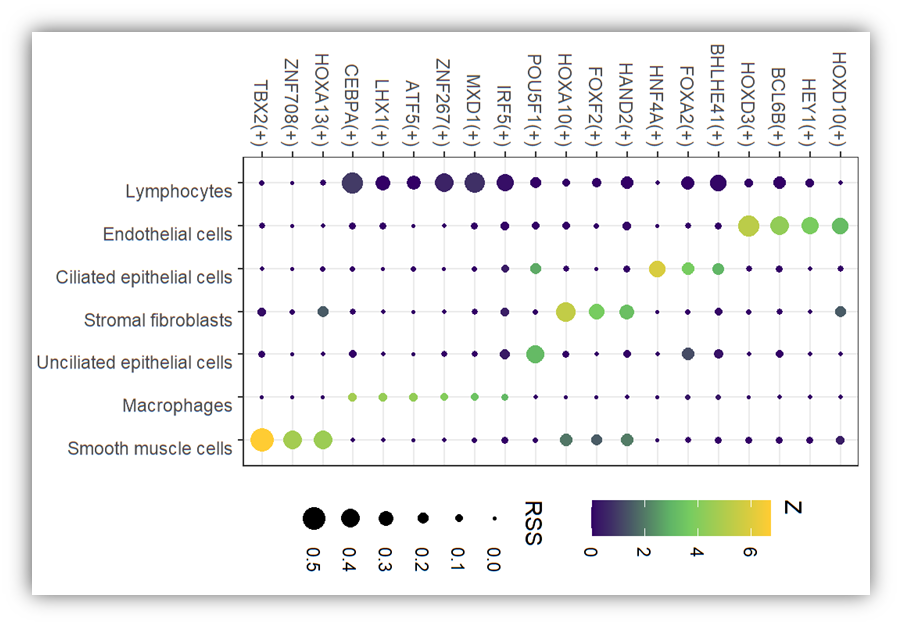
**pySCENIC全部往期精彩系列首先说一句,我们之前也发过R语言版本的SCENIC,但是后来我们感觉容易出错,而且费时,所以就没有再探究过。可是总是有小伙伴喜欢跑R,然后说这里错了,那里找不见,其实我们的帖子写于2022年,但是数据库已经更新了,去官网下载新的数据库,不能无脑跑代码。回到pySCENIC,之前我们写过整个系列4篇帖子,分析可视化都是很完善了。可是近期跑的时候发现在第一步有点问题
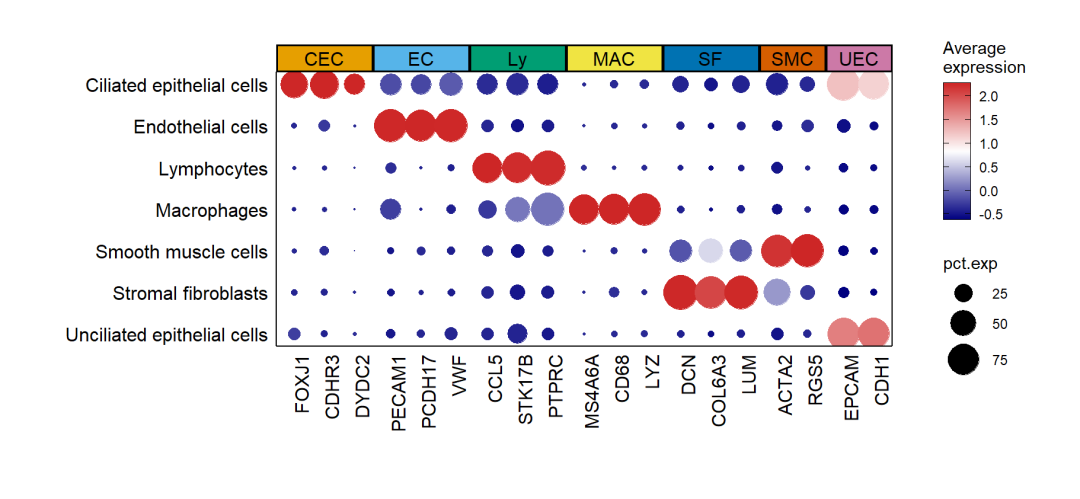
今天接着单细胞文章的内容:从Cell学单细胞转录组分析(一):开端!!!跟着Cell学单细胞转录组分析(二):单细胞转录组测序文件的读入及Seurat对象构建跟着Cell学单细胞转录组分析(三):单细胞转录组数据质控(QC)及合并去除批次效应跟着Cell学单细胞转录组分析(四):单细胞转录组测序UMAP降维聚类跟着Cell学单细胞转录组分析(五):单细胞转录组marker基因鉴定及细胞群注释前面几
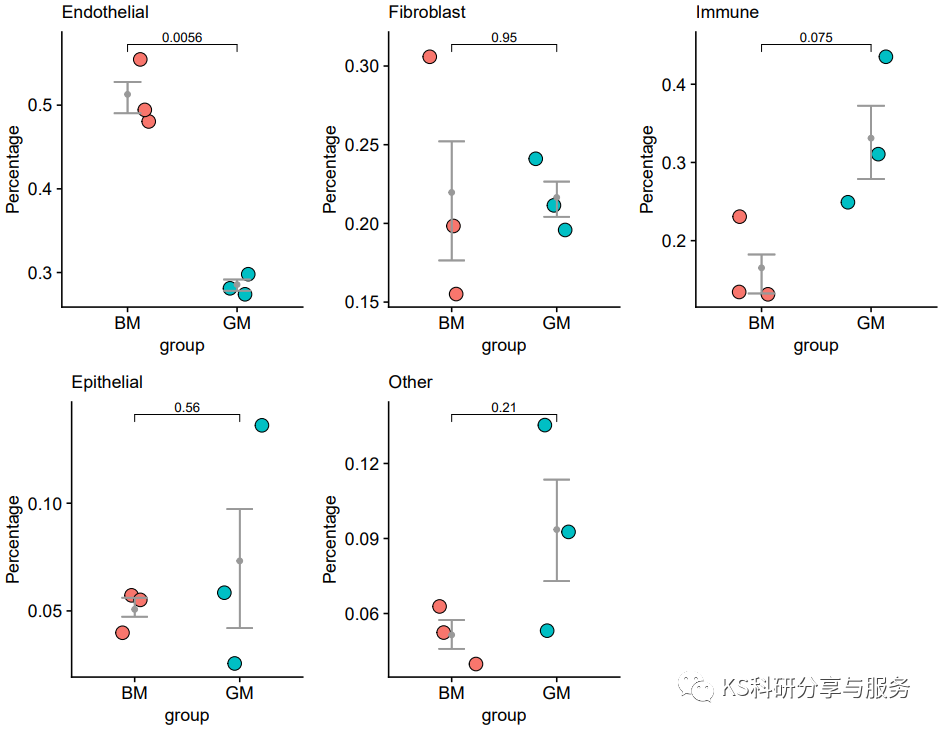
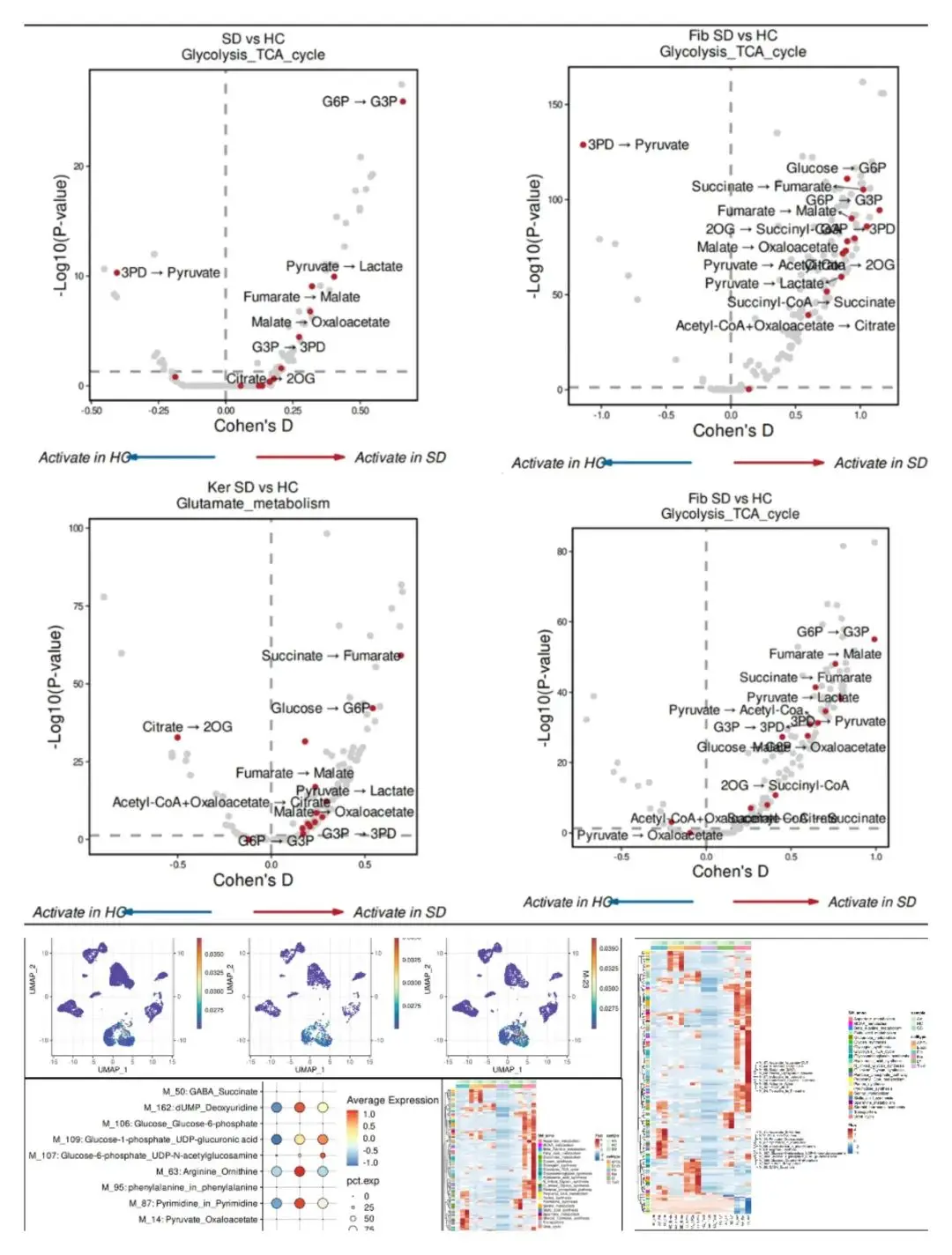
这次要介绍的scFEA(单细胞通量估计分析)也是基于单细胞转录组进行分析的。scFEA由人类代谢图谱作为重点代谢模块、利用scRNA-seq数据通量平衡约束的新型概率模型和基于图神经网络的新型优化求解器提供支持。利用多层神经网络捕获了从转录组到代谢组的复杂信息级联,以服从酶基因表达与反应速率之间的非线性依赖关系。具体原理感兴趣的可以读文章!我们演示了它的分析流程,以及一些下游可视化分析!相比于sc
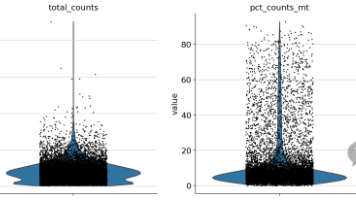
摘要:本文介绍了使用scanpy进行单细胞转录组数据质控的流程。主要内容包括:1)数据读取(10X数据格式);2)基础过滤(每个细胞至少表达200个基因,每个基因在至少3个细胞中表达);3)计算关键质控指标(线粒体基因、核糖体基因和血红蛋白基因的表达比例);4)可视化展示质控指标分布;5)根据实际数据情况设定过滤阈值(如线粒体基因比例<20%,总UMI数在1000-20000之间等)。文章强
这里分享一种单细胞数据可视化的图形修饰。先加载一个seurat公共的单细胞数据集InstallData("pbmc3k")data("pbmc3k")PBMC <- pbmc3k.finalPBMC = UpdateSeuratObject(object = PBMC)一般展示marker基因用这种点图:Markers <- c('AGER','SCGB3A2','TPPP3','CD

这里分享一种单细胞数据可视化的图形修饰。先加载一个seurat公共的单细胞数据集InstallData("pbmc3k")data("pbmc3k")PBMC <- pbmc3k.finalPBMC = UpdateSeuratObject(object = PBMC)一般展示marker基因用这种点图:Markers <- c('AGER','SCGB3A2','TPPP3','CD
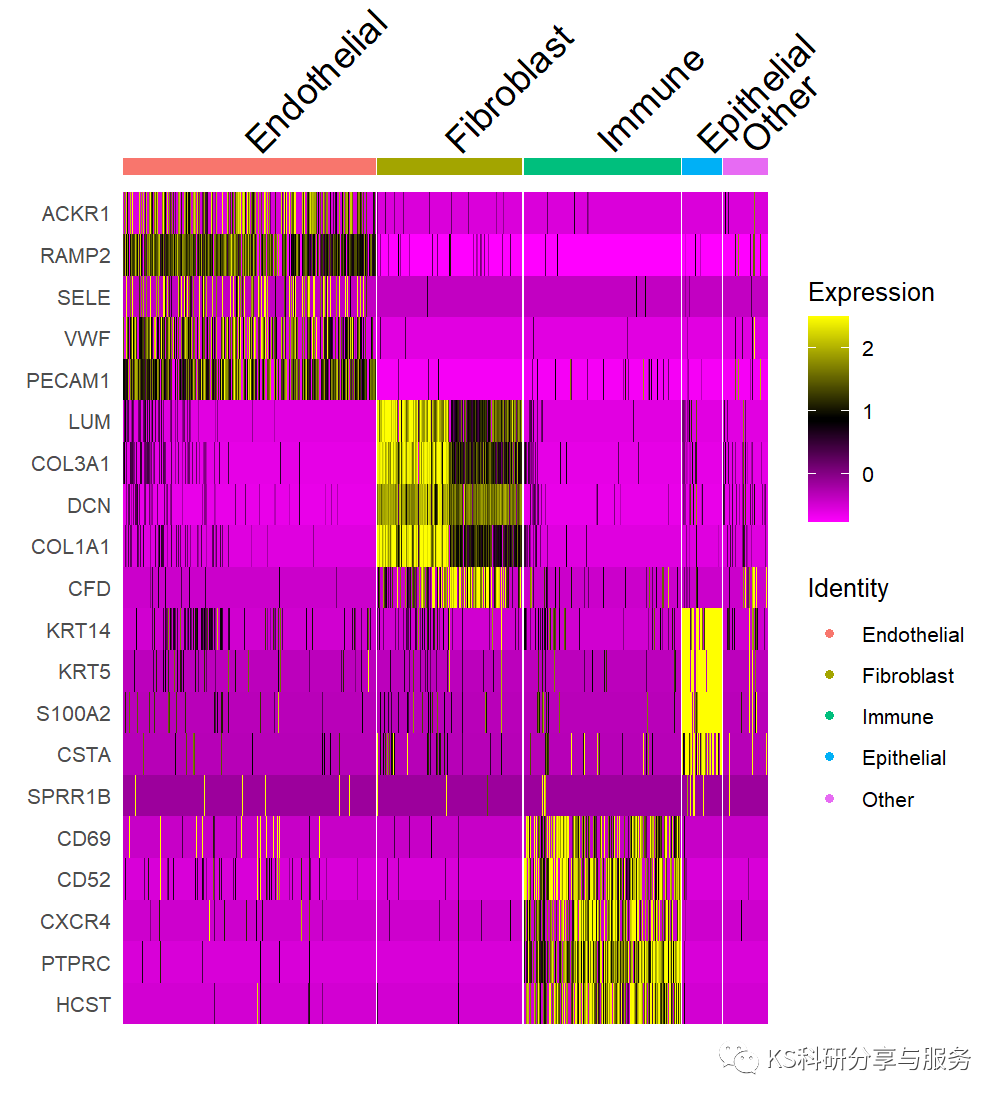
热图不再过多介绍了,参考之前的内容(热图系列大全)。单细胞基因可视化中热图也是比较受欢迎的,在分析完每群的marker基因之后,可以挑选显著的gene用seurat自带函数DoHeatmap可视化。当然也可以选任意自己想展示的基因进行可视化。首选选择基因,将其转化为列表,然后比对到原数据。markers <- c("ACKR1","RAMP2","SELE","VWF","PECAM1","
今天我们来复现一下一篇《cell》文章中的图标,图如下:(Liver Immune Profiling Reveals Pathogenesis and Therapeutics for Biliary Atresia)我们重点复现左边部分,右侧的作图在单细胞系列提到过:跟着Cell学单细胞转录组分析(十一):单细胞基因评分|AUCell评分,这里的复现不仅适用于单细胞,其他的数据也是可以做这样的

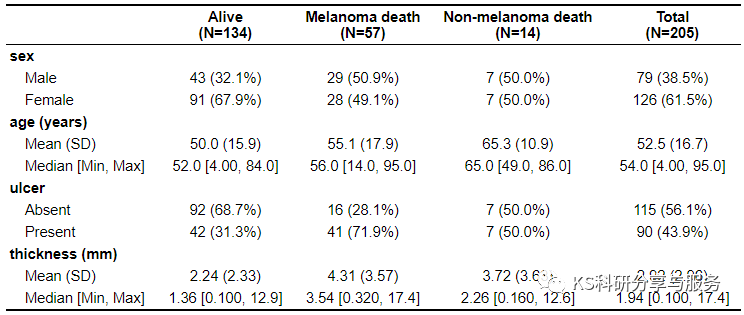
描述统计是统计学重要的一部分内容,尤其是在医学中的应用更广。很多医学、或者做统计的SCI文章,开头就是统计的内容,一般是由三线表的方式呈现的。之前小编也只会用excel统计然后自己制作表格,但这样效率很慢。这里我们介绍一个R包---table1,可以非常简单快速的完成统计工作,并制出三线表!安装包和示例数据install.packages("table1")install.packages("bo
,很多小伙伴购买函数,在使用过程却出现问题,主要的原因是我们在帖子里写的不够清楚,小伙伴也没有理解代码的意思,鉴于一个一个解释太费时间,我们决定写函数的帖子一律录制视频解说(视频在B站,搜索:KS科研分享与服务),方便大家使用。接下来看看bulk的效果,对于bulk表达数据,bulk基因表达量那一列列名要修改命名成exp,对于非单细胞的数据, 首先要进行设置的一个参数就是single_type=F