




- @kanghua_du
简介
该用户还未填写简介
擅长的技术栈
可提供的服务
暂无可提供的服务
本文介绍了生信分析中推荐使用服务器的原因及优势。作者指出,个人电脑虽可完成部分分析,但在处理单细胞、基因组等复杂数据时性能不足。自配服务器存在电费、运维等隐性成本,而云服务器(如西柚云)则提供了便捷、高性能的解决方案,无需设备维护,支持开具发票报销。文章还提供了西柚云服务器的优惠链接和操作指南,帮助用户快速选择适合的配置。
此推文是一个吐槽的推文,大家可以抱着吃瓜的心态观看。其实,自己并不想发次牢骚,但是,开源宇宙开源宇宙GPU oculink(目前);开源宇宙GPU服务器工厂)的做法实在让消费者难看。开源、拓展坞之类的产品,不可否认,他们在“开源”这块做的确实不错。自己也是很早以前就知道他们,也关注过他们的产品。此外,也购买了他们的oculink拓展坞。当时,我们的也就此出了学生及科研人员电脑配置推荐 | 笔记本+
这个教程也是前面《转录组上游分析》系列教程的中内容,我们今天单独的给作为一个章节来推送出来,因为,并不是所有人都是有这个系列教程有需求,可能你只是对某一个章节的内容感兴趣呢?
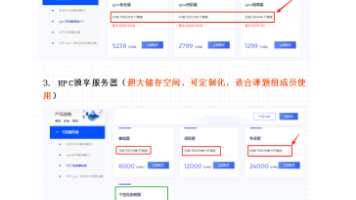
生物信息学分析推荐使用服务器而非个人电脑,主要原因包括:1)个人电脑仅能完成部分基础分析,如转录组等;2)高性能分析(单细胞、基因组等)需要大内存和计算资源;3)自建服务器存在运维、电费等隐性成本(如800W电源年电费约3504元);4)云服务器(如西柚云)提供便捷、高性能的解决方案,无需维护且网络访问快。建议根据分析需求和预算选择合适的计算资源,课题组无服务器的可优先考虑云服务方案。
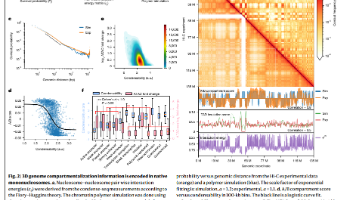
原文:Native nucleosomes intrinsically encode genome organization principles文章链接:https://www.nature.com/articles/s41586-025-08971-7#code-availability期刊:NatureIF:50.5发表时间:2025年5月7日Python。
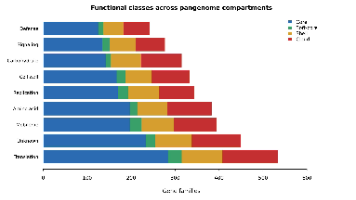
本文分享了一套基于Codex API开发的泛基因组分析流程代码,适用于细菌基因组比较分析。流程包含两种入口:复现文献公开基因组数据(使用NCBI数据)或处理新测序项目(从原始FASTQ开始)。主要步骤包括:软件环境配置(conda/mamba)、数据下载、质控修剪、基因组组装与矫正、功能注释和泛基因组分析(使用Panaroo/Roary)。项目提供了完整的脚本和R分析代码,支持可视化核心/附属基因
推荐大家购买最新的教程,若是已经购买以前WGNCA教程的同学,可以在对应教程留言,即可获得最新的教程。(注:此教程也仅基于自己理解,不仅局限于此,难免有不恰当地方,请结合自己需求,进行改动。删除~/.codex路径下已存在的config.toml文件(若有),然后新建一个config.toml,一是使用原生态的gpt模型,另外一种是使用国内其他大模型。创建好后,出现对应的PAI码,注意:此码仅出现
推荐大家购买最新的教程,若是已经购买以前WGNCA教程的同学,可以在对应教程留言,即可获得最新的教程。(注:此教程也仅基于自己理解,不仅局限于此,难免有不恰当地方,请结合自己需求,进行改动。删除~/.codex路径下已存在的config.toml文件(若有),然后新建一个config.toml,一是使用原生态的gpt模型,另外一种是使用国内其他大模型。创建好后,出现对应的PAI码,注意:此码仅出现
原文:Native nucleosomes intrinsically encode genome organization principles文章链接:https://www.nature.com/articles/s41586-025-08971-7#code-availability期刊:NatureIF:50.5发表时间:2025年5月7日Python。
国家水稻数据中心是中国水稻研究所创建的一个以水稻为核心,服务于育种需求的大型数据库,数据包含种质、突变体、分子标记、基因、QTL、文献资源等。,主要发表或收录生物信息学的教程,以及基于R的分析和可视化(包括数据分析,图形绘制等);Oryzabase是一个综合性水稻科学数据库,该数据库最初旨在收集尽可能多的信息.水稻基因组注释项目数据库(RGAP),主要提供水稻基因组序列和注释数据。**5. 水稻遗
