- @sweet_yemen
简介
该用户还未填写简介
擅长的技术栈
可提供的服务
暂无可提供的服务
在学习1中粗略地运行了一下软件的例子文件,但其中的很多东西都未能理解。该文中主要是对helper模块中代码的初步注释及学习以求能够熟练使用该软件。

可以看到,所有序列都存在一个“GTGTCAAATACTTATTTTCCCCGCTGTA”的前导序列,这可能是接头序列之类的,我们使用cutadapt工具将其去除。(注意:在使用samtools对bam文件进行索引之前必须对bam文件进行排序,否则会报错)不行,还是太大了,文件超过了三千万行,远超过了excel的处理能力,寻找其他方法进行统计。这是经公司使用fastp质控后的数据,我们先挑选部分数据
在学习1中粗略地运行了一下软件的例子文件,但其中的很多东西都未能理解。该文中主要是对helper模块中代码的初步注释及学习以求能够熟练使用该软件。
我们将介绍加载模型和预测突变影响的基本函数。
这个文章是学习与踩坑记录,包括一些处理流程以及遇到的坑的解决方法。
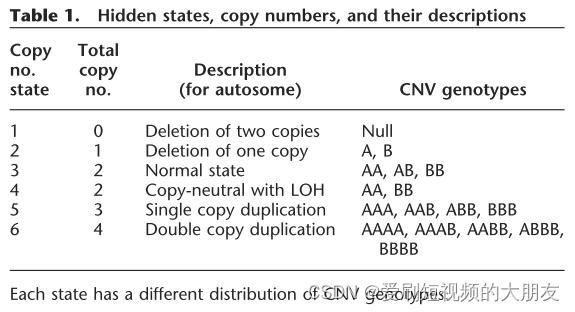
个体的杂合率偏差表明样品可能受到污染以及可能存在近亲繁殖。
再点进去之后就是每个芯片的分型数据,芯片的分型数据包含两个文件,即以Grn.idat和Red.idat结尾的原始分型文件, 即红光和绿光所在位置的测序文件。这个区域展示了SNP的信息,包括每个位点的染色体号,物理位置以及关键的每个个体在该位点识别的基因型。接着选择要分析的芯片的原始数据文件,注意这里只需要选择之前提到的以iDats结尾的文件夹即可。红框选择芯片对应的支持文件,这个文件可以在illu
第一步:进入DAVID网站后先择start analysis。1.选择upload2.在框框里粘贴要分析的目的基因集(也可以在下面以文件的形式上传)3.在Step 2:Select Identifier中选择与你上传的基因集的格式4.在Step 3:List Type中点第一个(就是要富集分析的基因集,第二个是支持自己上传背景基因集)5.在step4:Submit List中点击Submit Li