




- @m0_49960764
简介
该用户还未填写简介
擅长的技术栈
可提供的服务
暂无可提供的服务
物种内共线性分析步骤——JCVI安装以及数据下载(一)安装最简单的方法是通过PyPI安装它:1234pip install jcvi#或者安装开发版本pip install git+git://github.com/tanghaibao/jcvi.git或者,如果要手动安装:12git clone git://github.com/tanghaibao/jcvi.gitpip install...
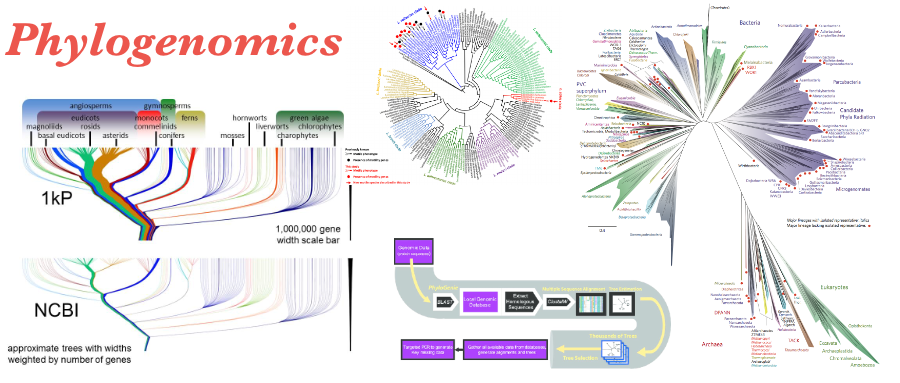
Phylogenomics写在前面关于系统发育基因组学的内容,本文参考了 Mike Lee 的文章,有一个相关的视频,时长为32‘51‘‘ 感兴趣的可以点击下方进入观看。系统基因组学:推断微生物之间的进化关系什么是系统基因组学?用一个容易理解但是不准确的概念来表示: 系统基因组学正试图在基因组水平上推断进化关系。因为在实践中,我们并没有关注的所有生物体整个基因组。并且根据
JCVI安装以及数据下载(MCScan)安装最简单的方法是通过PyPI安装它:pip install jcvi#或者安装开发版本pip install git+git://github.com/tanghaibao/jcvi.git或者,如果要手动安装:git clone git://github.com/tanghaibao/jcvi.gitpip install -e .还有一些依赖包安装方法
HIFI技术的简介HiFi reads(High fidelity reads)是Sequel II 三代测序平台推出的兼顾长读长和高准确度的测序序列,一般采用CCS(Circular Consensus Sequencing)模式测序。在这种测序模式下,酶读长一般大于插入片段长度,因此酶会绕着模板进行滚环测序,插入片段会被多次测序。单次测序中造成的随机测序错误,可以通过算法进行自我纠错校正,最终
写在前面上一篇介绍了 Snakemake 入门教程 做了一个简单的示例,具体查看我的上一篇内容下面会介绍一下 Snakemake的常用参数以及进阶的用法~介绍之前大家可以看一个视频了解一下(时长:19min14s, 选择性观看)Snakemake的简单介绍参数介绍命令行参数内核数调用$ snakemake --cores 1# 指定多个可用内核$ snake
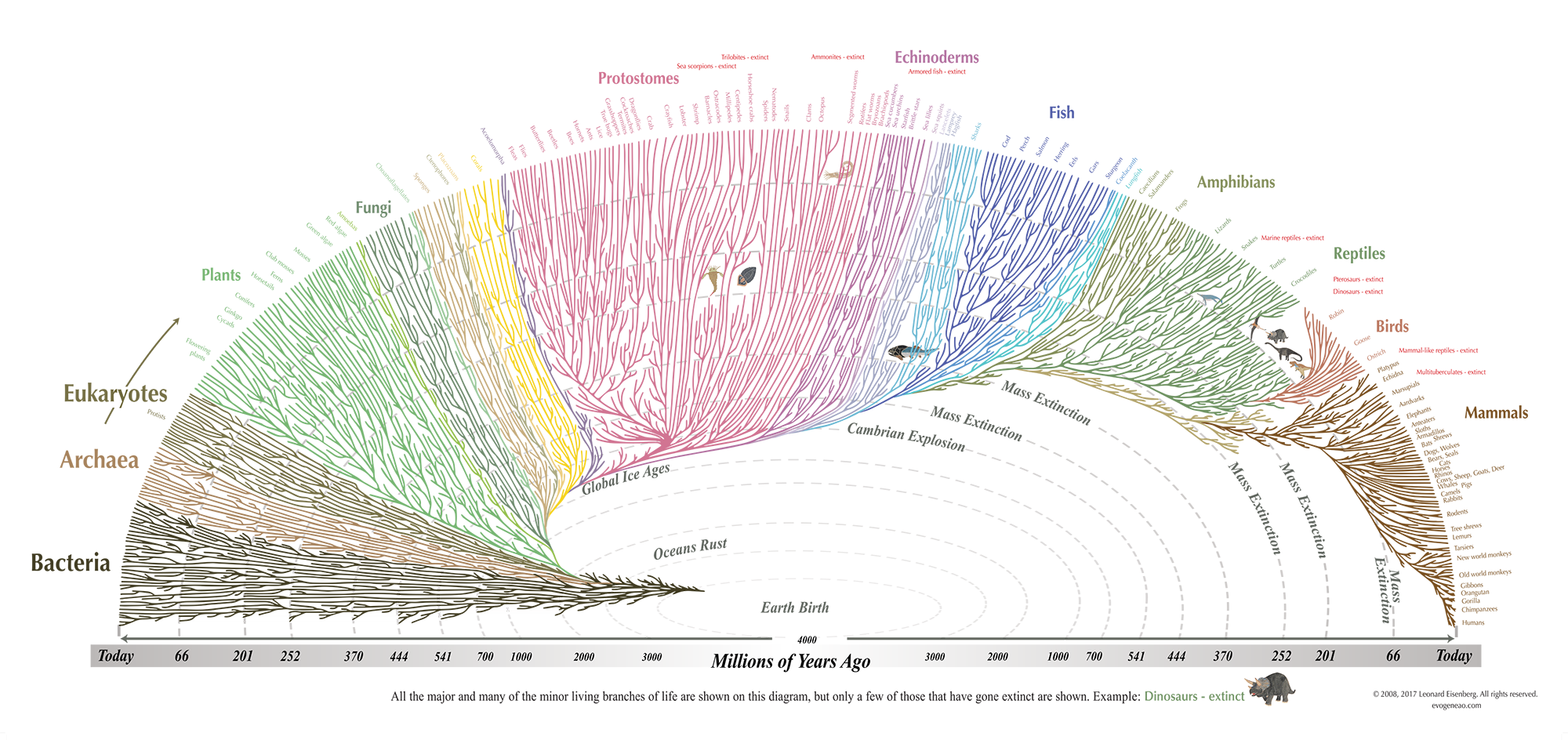
进化树构建进化树构建的问题是推断可能产生给定基因序列数据的进化树的拓扑结构和分支长度。推断树中叶节点的数量应等于给定数据中基因序列的数量。Neighbor-Joining AlgorithmNeighbor-Joining (NJ)树推理方法最初是由 Saitou 和 Nei 于 1987 年编写的。它属于一类基于距离的方法用于构建进化树。 NJ 方法采用给定序列之间的成对进化距离矩阵来构建进化树
title: 转录组分析流程|数据处理与De novo组装(一)tags:- 转录组组装- 教程- 软件- Trinity- Rcorrector- Trimmomaticcategories: 转录组分析转录组分析流程将分成三部分分别更新,更多内容关注我的个人博客定义转录组(transcriptome)广义上指某一生理条件下,细胞内所有转录产物的集合,包括信使RNA、核糖体RNA、转运RNA及非
物种内共线性分析(MCScanX+BLAST+TBtools)数据要求:做物种内共线性分析的话主要需要的是全基因组序列、cds或pep序列、gff3/gtf序列三者缺一不可。上一节下载好了cds序列以及gff3序列文件,以此为例(数据可在Phyzome下载,也可以在服务器上在线下载)软件要求:MCScanX、blast、TBtools(JCVI)物种内blast物种内blast 使用cds或p..