




- @GHZ2443063059
简介
该用户还未填写简介
擅长的技术栈
可提供的服务
暂无可提供的服务
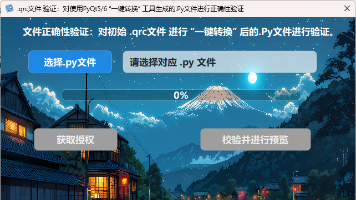
PyQtGHZ2025.1是一款专为PyQt5/PyQt6开发设计的工具,提供一键转换和验证功能,无需安装Python环境。主要功能包括:1)将.ui文件转换为可运行的.py窗体文件;2)将.qrc资源文件编译为*_rc.py;3)内置验证功能检查生成文件的可用性。4)一键使用 Qt Designer 免安装。该工具解决了PyQt开发中的环境配置复杂、命令行操作门槛高、版本兼容性等问题,特别适合教
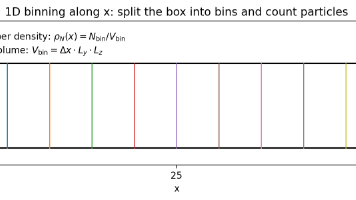
本文介绍了利用OVITO Python模块对分子动力学轨迹进行后处理分析的方法,重点讲解了如何计算沿特定方向的空间分布(如数密度、速度、应力等)。文章详细说明了直方图统计的核心原理,强调了后处理只能基于轨迹文件中已存在的原子属性。通过具体代码示例,展示了如何实现原子类型筛选、空间分箱、数密度计算以及多帧平均等操作,并指出了常见错误(如未设置fix_xrange、混淆计数与密度概念等)。对于需要统计
本文详细介绍了Gromacs进行蛋白-配体分子动力学模拟的完整流程。核心内容包括:1)所需软件环境配置(Gromacs、AmberTools等);2)模拟关键步骤(分子对接、拓扑文件生成、能量最小化、平衡模拟等);3)结果分析方法(结合能计算、氢键分析、RMSD/RMSF评估等)。文章特别强调了预处理的重要性,包括配体能量最小化和电荷平衡处理,并提供了PCA分析和自由能景观图绘制方法。最后指出可通

选择开源版:如果您主要进行日常的分子可视化和分析任务,并且希望使用免费的软件,开源版的 PyMOL 已经足够强大。它适合学术研究、教学以及个人项目,具有高度的可定制性。选择商业版:如果您从事的是更复杂的生物分子研究,或者需要生成高质量的图像和动画,并且希望获得官方的技术支持和更频繁的更新,那么商业版的 PyMOL 将是更合适的选择。尤其是在工业界和商业科研环境中,商业版的高级功能和优化能够显著提升

在分子动力学模拟中,MM/PBSA(分子力学-泊松-玻尔兹曼表面积)和 MM/GBSA(分子力学-广义玻恩表面积)方法被广泛用于估算生物分子复合物的结合自由能。针对 GROMACS 生成的轨迹文件,主要有两种工具可用于执行这些计算:gmx_mmpbsa.sh 和 gmx_MMPBSA。本文将详细比较这两者的特点、适用场景及各自优势,以帮助大家选择最适合的工具。

每日算法快闪赛是一种新兴的算法竞赛形式,具有时间短(15-60分钟)、题目精炼、反馈即时等特点。它为开发者提供了保持算法手感、模拟面试场景、拓展解题思路的机会,同时也能帮助团队进行技术内训和人才筛选。主流平台包括LeetCode、Codeforces等,配套工具涵盖题目推送、代码验证和可视化分析。参赛者需注重赛前准备、赛中策略和赛后复盘,组织者则需明确目标、选择合适题目并设计自动化流程。快闪赛未来

本文针对Mac电脑(M系列芯片)安装Schrodinger软件的特殊需求,提供了一套完整的安装方案。目前该软件在macOS平台安装面临三大难点:M芯片架构特殊、官方文档稀缺、依赖环境复杂。文章详细解析了从获取DMG安装文件、配置环境变量到License授权的全流程,并验证了Maestro、Glide、Desmond等核心模块的可用性。相比于Windows系统,macOS安装虽然存在技术门槛,但在命
在分子动力学模拟中,MM/PBSA(分子力学-泊松-玻尔兹曼表面积)和 MM/GBSA(分子力学-广义玻恩表面积)方法被广泛用于估算生物分子复合物的结合自由能。针对 GROMACS 生成的轨迹文件,主要有两种工具可用于执行这些计算:gmx_mmpbsa.sh 和 gmx_MMPBSA。本文将详细比较这两者的特点、适用场景及各自优势,以帮助大家选择最适合的工具。