
超详细机器学习(LASSO)教程,此时不学何时学?
在AI高速发展的时代,作为人工智能的一个重要分支,机器学习的热度大家有目共睹。要是能够在文献里用上一二,对论文质量提升较大。我们将介绍使用LASSO和随机森林(RandomForest)筛选特征基因。本期介绍LASSO。
在AI高速发展的时代,作为人工智能的一个重要分支,机器学习的热度大家有目共睹。要是能够在文献里用上一二,对论文质量提升较大。我们将介绍使用LASSO和随机森林(RandomForest)筛选特征基因。本期介绍LASSO。
小短文了解LASSO
一、LASSO 是什么?
LASSO 的全称是 “Least Absolute Shrinkage and Selection Operator”(最小绝对收缩与选择算子),是一种带 “正则化” 约束的回归方法。简单说,它的核心功能有两个:
-
收缩(Shrinkage):让不重要的特征系数 “变小”
-
选择(Selection):让一部分特征的系数直接 “变成 0”,相当于把这些特征从模型中 “剔除”
这种 “一边建模、一边筛选特征” 的能力,让它在高维数据(比如基因表达数据,特征数远多于样本数)中特别实用。
二、LASSO 的核心原理:L1 正则化
传统的线性回归(或逻辑回归)在特征太多时,容易出现 “过拟合”(模型太复杂,只记住了训练数据的细节,泛化能力差)。而 LASSO 通过加入一个 “L1 正则化” 约束,解决了这个问题。
通俗理解:
假设我们要通过 1000 个基因(特征)预测疾病状态(比如 “病例组” vs “对照组”),传统回归可能会给每个基因分配一个系数,但很多系数可能是 “噪音”(其实和疾病无关)。
LASSO 会在模型训练时加入一个 “惩罚项”:惩罚系数绝对值的总和(这就是 L1 正则化)。这个惩罚会 “迫使” 不重要的特征系数被 “压缩” 到 0—— 系数为 0,意味着这个特征对模型没有贡献,相当于被 “筛选掉” 了。
最终留下的,就是系数非零的 “关键特征”(比如与疾病相关的核心基因)。
三、LASSO 为什么适合基因数据筛选?
在生物信息学分析中,我们常遇到这样的情况:样本量较少、特征较多
这时候用传统回归会有两个麻烦:
-
特征太多,模型复杂到 “学不透”(过拟合)
-
无法判断哪些特征(基因)是真正和疾病相关的(传统回归不会主动剔除特征)
而 LASSO 的优势正好能解决这两个问题:
-
它能在 “控制模型复杂度” 的同时,自动把无关特征的系数压缩到 0,直接完成 “特征筛选”
-
最终留下的特征少而精,模型更简单、解释性更强(比如筛选出 3-5 个核心基因,比 “用 1000 个基因建模” 更容易理解)
四、关键参数:lambda(λ)—— 控制筛选 “严格度”
LASSO 的核心是一个叫 “lambda(λ)” 的参数,它决定了 “惩罚力度”:
- λ 越小:惩罚越轻,模型保留的特征越多(可能保留噪音,过拟合风险高);
- λ 越大:惩罚越重,模型会 “剔除” 更多特征(可能把有用的特征也删掉,欠拟合风险高)。
因此,选对 λ 是 LASSO 分析的关键。实际中,我们常用 “交叉验证”(比如 10 折交叉验证)来确定最优 λ:通过多次分割数据、训练模型,找到 “预测误差最小” 时对应的 λ(称为lambda.min),这个 λ 能平衡 “筛选精度” 和 “模型泛化能力”。
五、总结:LASSO 的核心价值
简单说,LASSO 就像一个 “智能筛选器”:面对成百上千的特征(比如基因),它能自动挑出那些 “真正有价值” 的变量,同时简化模型、避免过拟合。这也是为什么在基因表达、蛋白质组学等 “特征多、样本少” 的场景中,LASSO 几乎是特征筛选的 “标配” 工具。
接下来,我们就通过具体代码,看看如何用 LASSO 在数据中筛选与疾病相关的关键特征。
代码流程
-
1.基础工具准备(加载 R 包)
-
2.数据获取与初步观察
-
3.数据格式适配模型要求
-
4.模型构建与初步可视化
-
5.交叉验证优化关键参数
-
6.最终特征提取与结果保存
1. 加载分析所需的R包
library(tidyverse)#数据处理与可视化的综合包
library(glmnet)#用于拟合LASSO回归模型
library(e1071)#包含多种机器学习辅助函数2. 数据准备与初探
这里先看一下我们准备的数据,使用excel的转置功能整理表达矩阵得到
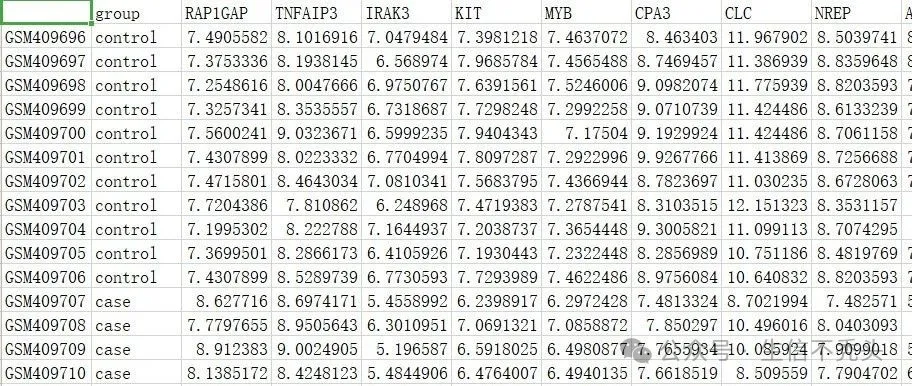
第一列为样本名,第二列为样本信息,后面的所有列为基因表达数据
# 设置工作目录(根据实际情况修改路径)
setwd("D:/文献复现_范科尼贫血/7lasso")
# 读取CSV格式的输入数据
# 参数说明:
# - row.names = 1:将第一列作为行名(通常是样本ID)
# - as.is = FALSE:保持数据原有类型,不自动转换
train <- read.csv("lasso准备数据.csv", row.names = 1, as.is = FALSE)
# 查看数据集基本信息
dim(train) # 查看数据集维度(样本数×特征数)
[1] 32 151
train[1:4, 1:4] # 查看前4行、前4列内容,了解数据格式
group RAP1GAP TNFAIP3 IRAK3
GSM409696 control 7.490558 8.101692 7.047948
GSM409697 control 7.375334 8.193815 6.568974
GSM409698 control 7.254862 8.004767 6.975077
GSM409699 control 7.325734 8.353556 6.731869这里通过维度可以看到,32行151列,因为包含1列样本信息,所以有32个样本与150个基因。
3. 数据预处理(适配LASSO模型)
# ----------------------------
# 提取特征矩阵(排除第一列分组信息)并转换为矩阵格式
# LASSO模型要求输入的特征必须是矩阵形式
x <- as.matrix(train[, -1])
# 处理因变量:将分组信息(group)转换为二元变量
# control组赋值为0,病例组赋值为1(便于二分类回归分析)
y <- ifelse(train$group == "control", 0, 1)
y # 查看转换后的因变量4. LASSO模型拟合与系数可视化
# 设置随机种子,确保结果可重复
set.seed(1314520)这里大家一定要养成设置随机种子的习惯。机器学习代码存在一定随机性(后面会提到)。设置随机种子可以确保结果的可重复性。例如从1到10中这10个数字抽取一个数字 两次,只要抽之前设置了相同的随机种子,那两次抽样得到的数字就是相同的。
# 拟合LASSO回归模型
# 参数说明:
# - x:特征矩阵
# - y:二元因变量
# - family = "binomial":二分类逻辑回归
# - alpha = 1:指定为LASSO回归(alpha=0为Ridge回归)
# - lambda = NULL:让模型自动选择lambda值范围
fit <- glmnet(x, y, family = "binomial", alpha = 1, lambda = NULL)
print(fit)
Call: glmnet(x = x, y = y, family = "binomial", alpha = 1, lambda = NULL)
Df %Dev Lambda
1 0 0.00 0.43810
2 2 5.91 0.41820
3 2 11.56 0.39920
4 2 16.69 0.38100-
Df(Degree of Freedom):当前Lambda下,非零系数的特征数量(即 LASSO 筛选出的 “有效特征数”)。
-
%Dev(Percentage of Deviance Explained):模型解释的偏差百分比。偏差(Deviance)是衡量模型拟合度的指标,%Dev越高,说明模型对 “因变量(如疾病分组)变异” 的解释能力越强。
-
Lambda:LASSO 的惩罚参数,λ 越大,惩罚越强(会更严格地压缩系数,甚至将系数逼到 0,减少特征数);λ 越小,惩罚越弱(系数更难被压缩,特征数易增加,模型拟合度更高)。
# 绘制LASSO回归系数路径图
# 展示不同lambda值下各特征系数的变化趋势
# 参数说明:
# - xvar = "lambda":x轴为lambda的对数变换值
# - label = TRUE:显示特征标签
plot(fit, xvar = "lambda", label = TRUE)
# 可选:以偏差解释率为x轴的系数路径图
# plot(fit, xvar = "dev", label = TRUE)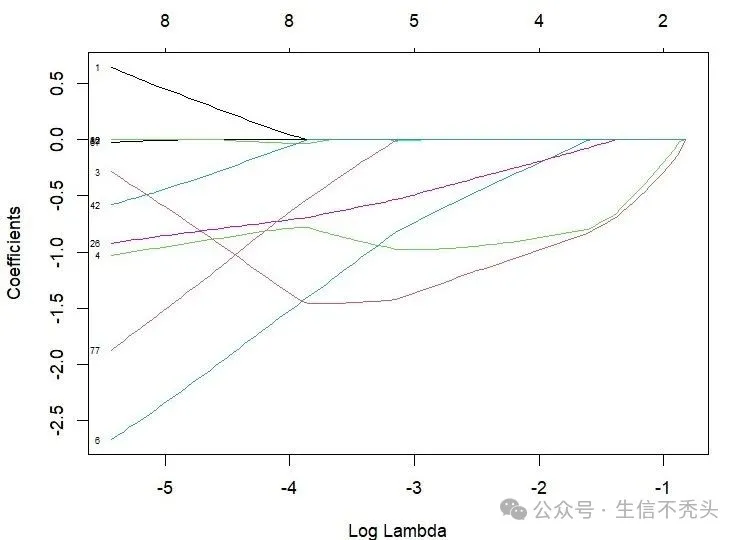
解读:
-
横轴(Log Lambda):λ 是 LASSO 的惩罚参数,λ 越大(Log Lambda 越靠左,如 - 5 附近),惩罚越强;λ 越小(Log Lambda 越靠右,如 - 1 附近),惩罚越弱。
-
纵轴(Coefficients):回归系数,表示 “特征与因变量(如疾病分组)的关联强度和方向”(正系数 = 正相关,负系数 = 负相关,绝对值 = 关联强度)。
这张图直观展示了:LASSO 如何通过调整惩罚强度(λ),从 “所有特征” 中逐步筛选出 “与因变量最相关的关键特征”—— 惩罚越强,筛掉的特征越多;惩罚越弱,保留的特征越多,每条彩色曲线代表一个特征(基因)。
5. 交叉验证确定最佳lambda
# 进行10折交叉验证,优化lambda值
#对于该数据来说,可能5倍折叠更好,但按照文献,我们还是选择的10倍折叠
# 参数说明:
# - nfold = 10:10折交叉验证
# - type.measure = 'deviance':以偏差作为评价指标
cvfit <- cv.glmnet(x, y, nfold = 10, family = "binomial", type.measure = 'deviance')
# 可选:以分类错误率作为评价指标
# cvfit = cv.glmnet(x, y, nfold=10, family = "binomial", type.measure = "class")这里的nfold折叠数需要特别注意,简单来说nfold指的是将原始数据集分割成的若干个 “子集合”(这里存在随机性)。然后用部分数据训练模型,用另一部分数据验证模型。
为什么要 “分折叠”?
核心是为了 让模型评估更客观。
-
如果不分割折叠,直接用 “全部数据训练,再用全部数据验证”,模型很可能 “记住” 了训练数据的细节(过拟合),导致评估结果虚高(看起来很好,但实际预测新数据时很差)。
-
而 “分折叠” 通过 “训练用大部分数据,验证用小部分新数据”,能更真实地模拟模型在 “未见过的数据” 上的表现,避免过拟合带来的误导。
下方表格仅供大家参考!折叠数还需根据具体样本信息进行选择
|
样本数 |
推荐折叠数 |
核心原因 |
|
样本量较小(<50) |
5 折 |
避免验证集过小导致的结果波动,提升评估稳定性 |
|
样本量较大(50~200) |
10 折 |
平衡偏差与方差,结果稳定,符合领域惯例 |
# 绘制交叉验证结果图
# 展示不同lambda值对应的交叉验证误差
# 图中虚线分别表示最小误差点和1个标准差范围内的最佳lambda值
plot(cvfit)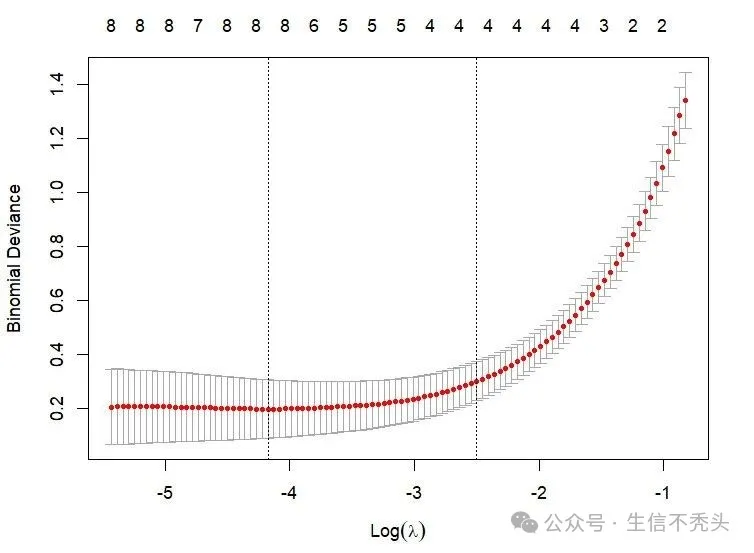
解读:
-
红色点线:不同 λ 对应的交叉验证平均偏差(即 10 折交叉验证后,模型在验证集上的平均误差)。
-
灰色竖线:偏差的标准差(表示 10 次交叉验证中,误差的波动范围)。
-
垂直虚线:通常有两条,对应两个关键 λ:
左侧虚线:λ.min(使交叉验证偏差最小的 λ,拟合效果最优)。
右侧虚线:λ.1se(在 λ.min 基础上,偏差增加不超过 1 个标准差的最大 λ,目的是选 “更简洁的模型”—— 惩罚更强,筛选的特征更少)。
-
上方数字:对应 λ 下非零系数的特征数量(即 LASSO 筛选出的 “有效特征数”)。
对于机器学习的结果图确实比较难懂,但论文写作时一般不会要求过多解读
6. 特征筛选与结果保存
# 查看交叉验证得到的最小误差对应的lambda值(最佳lambda)
cvfit$lambda.min
[1] 0.01538247
# 根据最佳lambda值提取模型系数
myCoefs <- coef(cvfit, s = "lambda.min")如何选最佳 λ?
-
追求 “最佳拟合”:选 λ.min(左侧虚线对应位置)—— 此时模型在验证集上误差最小,但可能保留较多特征。
-
追求 “简约模型”:选 λ.1se(右侧虚线对应位置)—— 此时偏差仅比最小值高 1 个标准差,但惩罚更强,筛选的特征更少(上方数字更小),模型更简洁且泛化能力可能更强。
这里我们选择lambda.min作为最佳λ
lasso_fea <- myCoefs@Dimnames[[1]][which(myCoefs != 0)]
lasso_fea <- lasso_fea[-1] # 排除第一个截距项(Intercept)
# 展示LASSO回归筛选出的关键特征
lasso_fea
[1] "RAP1GAP" "IRAK3" "KIT" "CPA3" "PRG2" "HSPA1A" "IGLL1" "VNN1"
# 将筛选得到的特征保存到CSV文件,用于后续分析
write.csv(lasso_fea, "3feature_lasso.csv")至此分析结束,以上内容均为个人学习经验,如有错误,欢迎大家反馈、讨论

欢迎加入北京社区
更多推荐



 27
27 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)