
Alphafold2源代码导读
Alphafold2源码解读
Alphafold2源代码导读

作者: 谷雨
链接:https://zhuanlan.zhihu.com/p/492381344
介绍主要涵盖的部分是:
- 特征数据搜索和特征提取/预处理
- 模型设置与Alphafold主网络架构的控制流
源代码架构
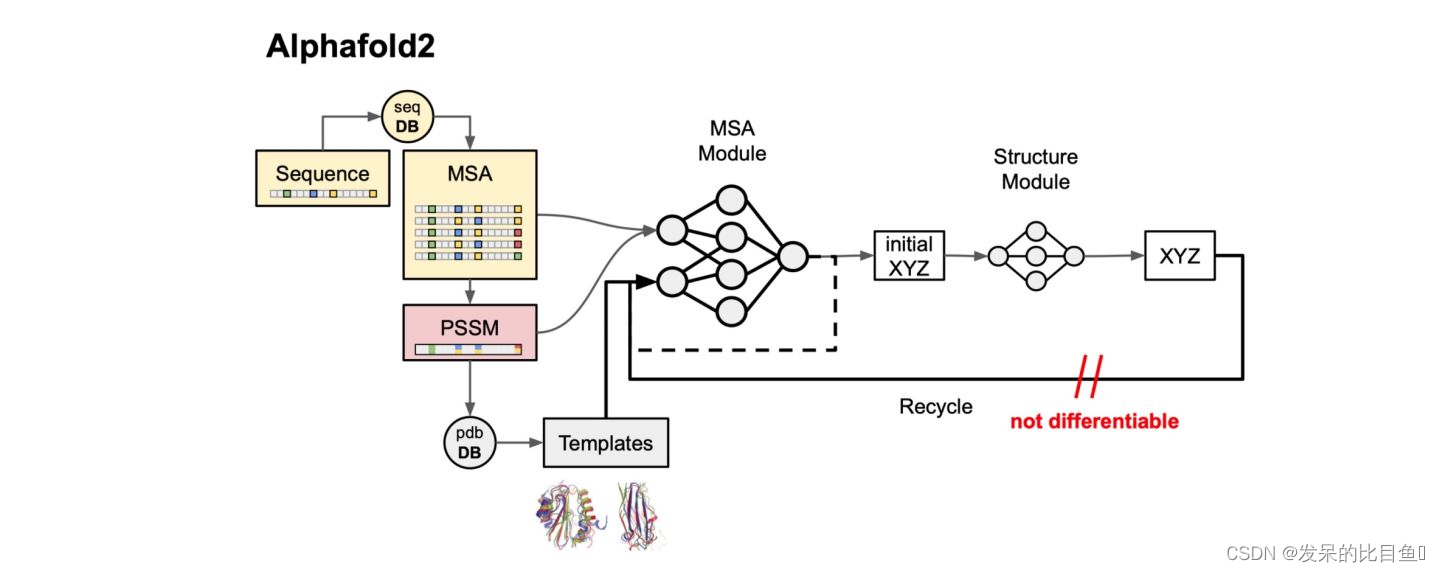
代码组层:
run_alphafold:顶层封装,负责传递参数和运算。
alphafold.common
- confidence:处理confidence metrics的函数
- protein:PDB处理蛋白表征的函数集合,比如from_pdb_string,to_pdb,from_prediction得到结构表征数据。
- residue_constants:储存数据常数的地方,比如residue_atoms、rigid_group_atom、 sequence_to_onehot等。
alphafold.data
- tools: MSA搜索的工具包如hhblits、hhsearch、jackhmmer等
- tf: 负责处理和转化raw特征为alphafold主干模型需要的所有input特征
- msa_identifiers:MSA预处理与特征提取
- templates:提取PDB结构模板特征提取
- pipeline:所有特征提取的处理流程模块,包括MSA搜索、template搜索、特征整合的Protocol模块
- parsers:读取和转化各类MSA、fasta相关文件的模块
- msa_pairing:处理multimer的msa pairing的模块
- feature_processing:multimer特征embeddings处理流程函数
alphafold.model:主要的模型编码储存在此
- modules_multimer:Multimer的Evoformer模块代码
- modules:Evoformer核心模块代码
- folding:structure module的核心代码
- quat_affine:structure module的四元数处理模块
- r3:rotation处理模块
- utils: jax的通用函数
- folding_multimer:multimer的structure module
- lddt:评估lddt的函数
- layer_stack:将模块堆叠的操作
- features:将预处理的特征进行tensor化的模块
- data:预训练模型参数loading的模块
- config:模型超参或option参数管理模块
- all_atom:原子表示的模块
- all_atom_multimer:multimer的原子表示的模块
- common_modules:定义了一个特殊的haiku Linear层。
- prng:haiku随机数处理的代码
alphafold.relax:amber relax的API
- amber_minimize:amber能量最小化
- cleanup:amber预处理
- relax:relax主程序
- utils:一些relax相关的通用函数
执行函数
最外层的run_alphafold.py其中main函数是主要的执行函数,简化流程示意图:
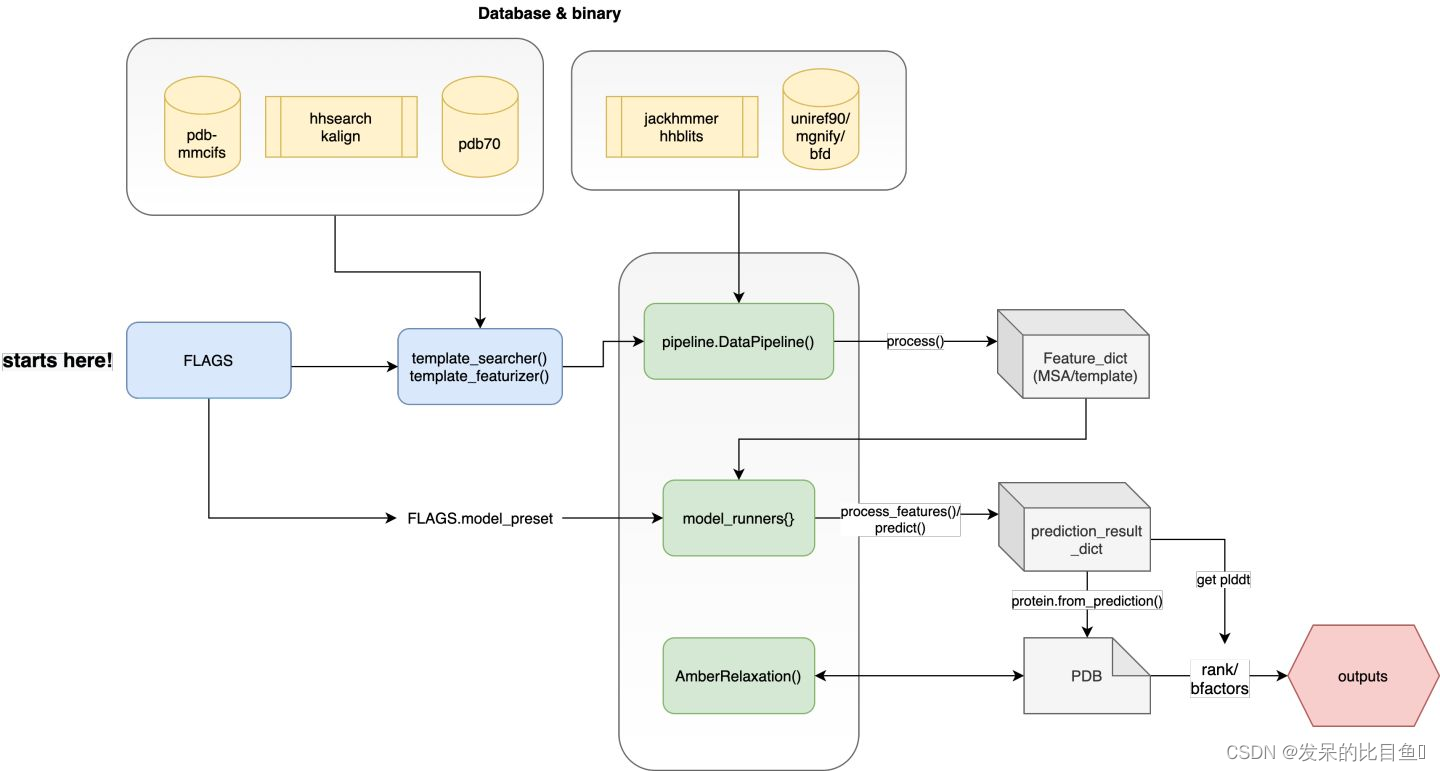
其主要定义了4块主要内容:
**Step1:**借助absl FLAG模块定义第三方程序路径、各类数据库的搜索路径以及各类运行参数;
**Step2:**根据DataPipeline流程,对MSA进行搜索、对模板进行搜索并构建所有需要的特征;
**Step3:**设置模型model_name, configs等信息,对haiku model container进行实例化和jit编译,process_feature():对特征字典的内容进行张量化;predict(): 运行alphafold主模型进行结构预测;
**Step4:**执行amber_relax, 对PBD坐标进行能量优化,打分排名和输出结果
absl.FLAG参数控制
run_alphafold.py是af2的主程序控制的部分,其使用了absl来管理python command line的参数。
路径控制部分:
- hhblits、jackhmmer、hhsearch、kalign等第三方MSA search程序的binary_path
- uniref90、bfd、mgnify、small_bfd、uniclust30、uniprot_database、pdb70、pdb_seqres、-obsolete_pdbs等路径
- fasta_paths:fasta文件的路径
- output_dir:输出数据的路径
- data_dir:model weights的路径
模型参数控制:
- model_preset: 预模型选择参数,可选[‘monomer’, ‘monomer_casp14’, ‘monomer_ptm’, ‘multimer’]
- db_preset:template数据库的类型,[‘full_dbs’, ‘reduced_dbs’]
- random_seed: 输入顶层初始的随机数种子
- use_precomputed_msas:是否使用预先计算好的msa, [False, True]
- run_relax/use_gpu_relax: 是否使用amber relax步骤
- benchmark: 跑分模式, [False]
- num_multimer_predictions_per_model 针对multimer模式,控制模型输出总数量
DataPipeline
主要负责处理MSA/Template的特征处理流程简图如下:
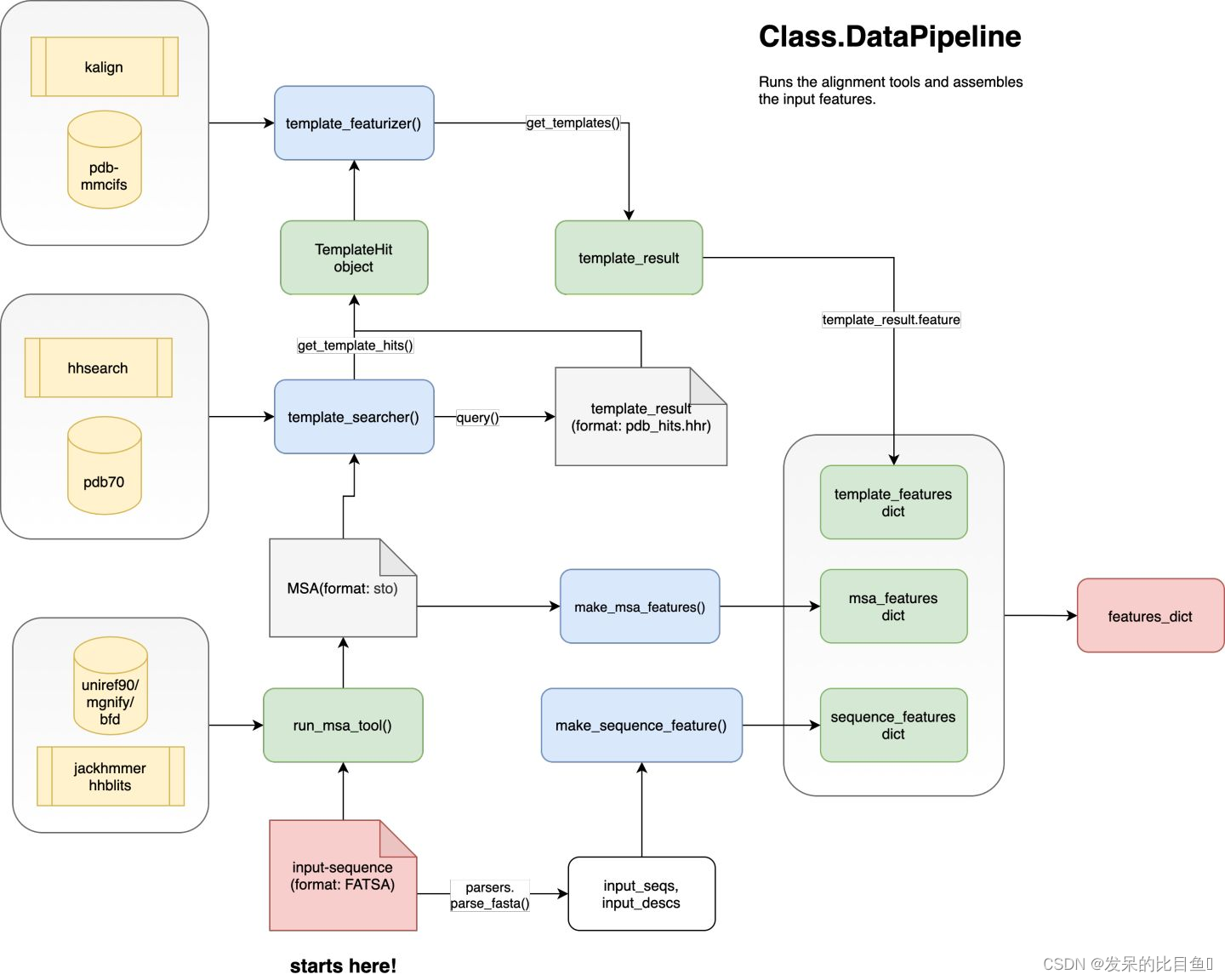
主要调动的API是:
- 通过使用run_msa_tool()函数调用jackhmer和hhblits对输入的Fasta文件进行MSA搜索,输出的格式为sto;
- template_searcher()使用去冗余后的MSA序列对pdb70数据库进行搜索,得到TemplateHits;
- template_featurizer()使用kalign程序对Hits的序列与input序列进行比对,得到模板信息;
- 最后通过make_*_features()函数对特征进一步处理,与融合得到alphafold所需的所有特征字典。
input sequence特征提取
对input的FASTA文件信息进行处理:
- aatype: one-hot编码的序列
- between_segment_residues?含义不明(可能在multimer的文章中会有解释)
- domain_name:序列名,字符串信息
- residue_index: 残基编号,从0开始
- seq_length:序列长度,为什么会重复N次?
- sequence:人类可读的序列3字母缩写序列,类型字符串
from alphafold.data.pipeline import make_sequence_features
# extraction:
sequence_features = make_sequence_features(
sequence=input_sequence,
description=input_description,
num_res=num_res)
-----------------------------------------------------------------------------
# Return Format:
{'aatype': array([[0, 0, 0, ..., 0, 0, 0],
[0, 0, 0, ..., 0, 0, 0],
...,
[0, 1, 0, ..., 0, 0, 0]], dtype=int32, shape=Lx21),
'between_segment_residues': array([0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0], dtype=int32, shape=L,),
'domain_name': array([b'4FAZA'], dtype=object),
'residue_index': array([ 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16,17, 18, 19,...50,51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61], dtype=int32, shape=L,),
'seq_length': array([62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62,62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62,
62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62, 62], dtype=int32,shape=L,),
'sequence': array([b'PFAQIYLIEGRTEEQKRAVIEKVTQAMMEAVGAPKENVRVWIHDVPKENWGIGGVSAKALGR'],
dtype=object)}
MSA特征提取
MSA的处理分为2个主要步骤:
- 使用parsers对msa的内容进行格式化:Msa object,其中有三个属性:
- sequences:同源序列的python list
- deletion_matrix: 检测MSA中每条序列中是否存在小写字符的氨基酸信息,此区域代表同源序列中的序列被删除。(在推理中貌似没用,矩阵的维度是NxL),可能的信息Training时在Residue cropping部分。
- descriptions:同源序列来源信息的python list
from alphafold.data.pipeline import make_msa_features
from alphafold.data import parsers
# parsers msa strings from a3m:
a3m_file = 'uniref.a3m'
with open(a3m_file, 'r') as f:
a3m_str = f.read()
# parse_a3m, 读取序列和deletion_matrix信息:
uniref90_msa = parsers.parse_a3m(a3m_str)
uniref90_msa.sequences
---------------------------------------------------------------
['PFAQIYLIEGRTEEQKRAVIEKVTQAMMEAVGAPKENVRVWIHDVPKENWGIGGVSAKALGR',
'PFAQIYMIEGRTEEQKKAVIEKVTQALVDAVGAPPANVRVWIHDVPKENWGIAGQTAKELGR']
jnp.array(uniref90_msa.deletion_matrix)
---------------------------------------------------------------
DeviceArray([[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0]], dtype=int32)
uniref90_msa.descriptions
---------------------------------------------------------------
['101', 'UPI00188DD275']
Msa Object包含的信息:
def parse_a3m():
...................
return Msa(sequences=aligned_sequences,
deletion_matrix=deletion_matrix,
descriptions=descriptions)
- 调用make_sequence_features()对MSA特征去重、MSA序列数字list化以及更新特征:
# extract MSA feartures
from alphafold.data.pipeline import make_msa_features
msa_features = make_msa_features([uniref90_msa])
msa_features.keys()
------------------------------------------------------------------
dict_keys(['deletion_matrix_int', 'msa', 'num_alignments', 'msa_species_identifiers'])
具体特征的含义:
# 将MSA每条序列转为以数字替代的list,方便后续one-hot化
msa_features['msa']
------------------------------------------------------------------
array([[12, 4, 0, 13, 7, 19, 9, 7, 3, 5, 14, 16, 3, 3, 13, 8,
14, 0, 17, 7, 3, 8, 17, 16, 13, 0, 10, 10, 3, 0, 17, 5,
0, 12, 8, 3, 11, 17, 14, 17, 18, 7, 6, 2, 17, 12, 8, 3,
11, 18, 5, 7, 5, 5, 17, 15, 0, 8, 0, 9, 5, 14],
[12, 4, 0, 13, 7, 19, 10, 7, 3, 5, 14, 16, 3, 3, 13, 8,
8, 0, 17, 7, 3, 8, 17, 16, 0, 0, 9, 17, 21, 21, 21, 21,
21, 21, 21, 21, 11, 17, 14, 17, 18, 7, 6, 2, 17, 12, 8, 3,
11, 18, 5, 7, 0, 5, 13, 16, 0, 8, 3, 9, 5, 14]],
dtype=int32, shape=N*xL)
msa_features['deletion_matrix_int'] = 去冗余后的deletion_matrix
------------------------------------------------------------------
array([[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0]],
dtype=int32)
msa_features['num_alignments'] = [num_alignments] * num_res
------------------------------------------------------------------
array([2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2,
2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2,
2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2, 2], dtype=int32)
msa_features['msa_species_identifiers'] 种属信息,貌似我提供的MSA中没有这部分东西
------------------------------------------------------------------
array([b'', b''], dtype=object)
Template特征提取
具体细节:参照alphafold2补充材料原文–1.2.3 Template search
- template_searcher使用HHSearch
template的搜索依赖于hhsearch、pdb70数据库,以及预处理好的MSA序列信息
from alphafold.data.tools import hhsearch
hhsearch_binary_path = '/usr/local/bin/hhsearch'
pdb70_database_path = '/content/drive/MyDrive/af2_backprop/fake_pdb70/pdb70' # need cif;
# searcher:
template_searcher = hhsearch.HHSearch(binary_path=hhsearch_binary_path, databases=[pdb70_database_path])
# 使用a3m数据类型进行:
pdb_templates_result = template_searcher.query(a3m_str)
------------------------------------------------------------------
pdb_templates_result是人类可读的字符,即hhsearch的输出,可以把这些及结果写入pdb_hits.hhr中。
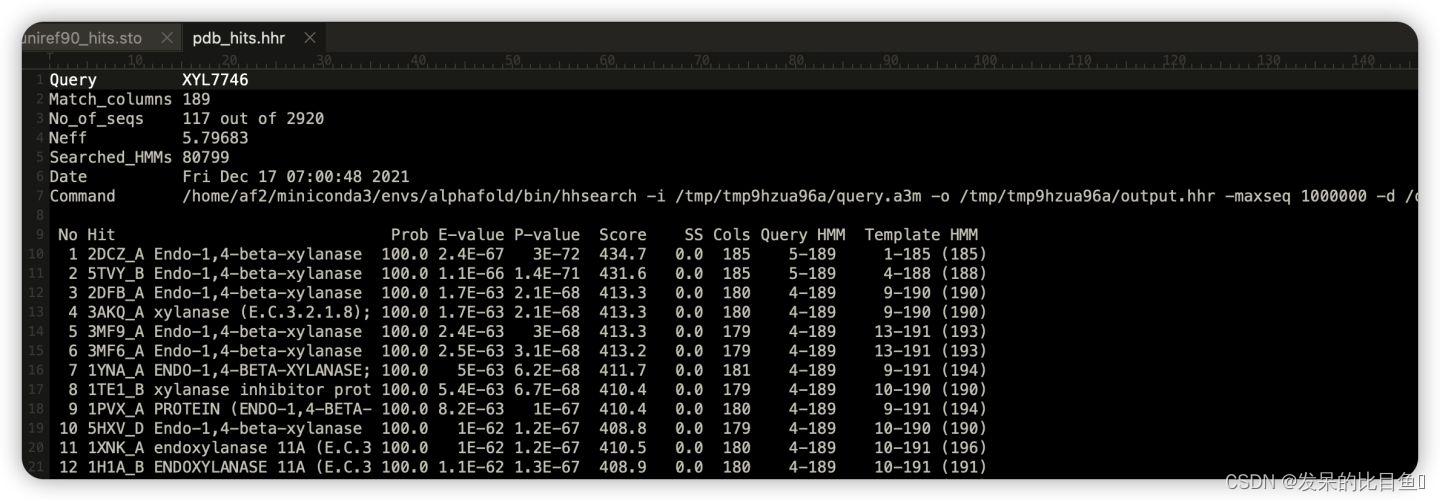
其中主要记录的是可用模板的PDB ID,序列相似度,结构与序列match的区间**
- 将pdb_hits.hhr中的信息格式
- aligned_cols:指query sequence被模板覆盖的区间大小
- sum_probs被用于对template进行打分排名
- indices_query,indices_hit是最重要的信息,这里记录了模板和query sequence的匹配区间信息
3. 提取template的特征信息
template特征提取部分参考alphafold2补充材料–1.2.9 Featurization and model inputs
- 此处可以根据PDB的release日期选择模板,方便测试等用途,由max_template_date参数控制,获取一定数量的模板进行后续处理;
- 使用biopython将query的序列和mmcif结构文件中的序列进行匹配对应,并确认残基或原子是否存在时,若存在,分离xyz坐标的信息并标记对应位置的mask=1;
- 根据sum_probs对模板进行排名,默认使用4个最好的模型进行特征提取。
from alphafold.data import templates
# template featurize:
max_template_date_ = '2022-01-01'
MAX_TEMPLATE_HITS = 20
kalign_binary_path_ = '/usr/local/bin/kalign'
obsolete_pdbs_path_ = None # 用于可自定义模板;
template_mmcif_dir = '/content/drive/MyDrive/af2_backprop/mmcif_db'
# featureizer:
template_featurizer = templates.HhsearchHitFeaturizer(
mmcif_dir=template_mmcif_dir,
max_template_date=max_template_date_,
max_hits=MAX_TEMPLATE_HITS,
kalign_binary_path=kalign_binary_path_,
release_dates_path=None,
obsolete_pdbs_path=obsolete_pdbs_path_)
# get features:
templates_result = template_featurizer.get_templates(query_sequence=input_sequence, hits=pdb_template_hits)
template的raw特征中包含什么?:
# keys:
templates_result.features.keys()
dict_keys(['template_aatype', 'template_all_atom_masks', 'template_all_atom_positions', 'template_domain_names', 'template_sequence', 'template_sum_probs'])
template的feature特征包括:N指模板的数量
- template_aatype: 模板序列的one-hot representation,shape = NxLx22,包括unknown和gap;
- template_all_atom_masks**: shape = NxLx37,代表在模板中,原子是否存在,存在=1,不存在=0;
- template_all_atom_positions**: shape = Lx37x3, 其中37为所有的可能的蛋白原子类型,3维代表xyz坐标值。
- template_domain_names: 模板的名称
- template_sequence: shape =NxL 序列字符串
- template_sum_probs: match的打分值 (np.float32)
至此,input_sequence, MSA_sequence, structure template的raw feature全部被提取(大多数都是one-hot类型的向量),这些特征需要被进一步处理为tensor以及衍生出更多的特征数据。
合并Feature_dict
# cat all features;
feature_dict = {**sequence_features, **msa_features, **templates_result.features}
RunModel
RunModel是Alphafold主干网络的容器,主要的代码存在于alphafold.model。
主要流程示意图:

模型设置
AlphaFold v2.2存在多个weights参数,所有的weights name可以在alphafold.model.config中查看:
from alphafold.model import config
config.MODEL_PRESETS
------------------------------------------------------------
{'monomer': ('model_1', 'model_2', 'model_3', 'model_4', 'model_5'),
'monomer_casp14': ('model_1', 'model_2', 'model_3', 'model_4', 'model_5'),
'monomer_ptm': ('model_1_ptm',
'model_2_ptm',
'model_3_ptm',
'model_4_ptm',
'model_5_ptm'),
'multimer': ('model_1_multimer_v2',
'model_2_multimer_v2',
'model_3_multimer_v2',
'model_4_multimer_v2',
'model_5_multimer_v2')}
并且config还存储了许多其他的alphafold的维度的预设参数: 比如记录data的维度或model的dropout_rate, attention的头数等等。
# look all configure:
config.CONFIG
config.CONFIG_MULTIMER
认真看部分的设置会发现,有一些模型并不使用template特征:
config.CONFIG_DIFFS

模型初始化
模型的初始化需要config和model name, 需要加载对应的模型参数以完成实例化。
from alphafold.model import config
# 储存模型的字典:
model_preset = 'monomer_ptm'
model_runners = {}
num_ensemble = 1
# get all weights name:
model_names = config.MODEL_PRESETS['monomer_ptm']
for model_name in model_names:
# 实例化每个模型的config:
model_config = config.model_config(model_name)
if run_multimer_system:
model_config.model.num_ensemble_eval = num_ensemble
else:
model_config.data.eval.num_ensemble = num_ensemble
# 加载模型weight:
model_params = data.get_model_haiku_params(
model_name=model_name, data_dir=FLAGS.data_dir)
# 实例化haiku model:
model_runner = model.RunModel(model_config, model_params)
# 储存在字典中:
for i in range(num_predictions_per_model):
model_runners[f'{model_name}_pred_{i}'] = model_runner
如果解析更底层一些可以看看RunModel的类,实例化的部分的主干代码在alphafold.model.modules.AlphaFold()
- batch: 指的是DataPipeline处理得到的feature_dict;
- hk.transform将jax model转换为可以被即时编译的纯函数,得到init() 和 apply()
from alphafold.model import modules
class RunModel:
"""Container for JAX model."""
def __init__(self,
config: ml_collections.ConfigDict,
params: Optional[Mapping[str, Mapping[str, np.ndarray]]] = None):
# configure
self.config = config
# weights
self.params = params
# use multimer?
self.multimer_mode = config.model.global_config.multimer_mode
if self.multimer_mode:
def _forward_fn(batch):
model = modules_multimer.AlphaFold(self.config.model) # 实例化部分
return model(
batch,
is_training=False)
else:
def _forward_fn(batch):
model = modules.AlphaFold(self.config.model) # 实例化部分
return model(
batch,
is_training=False,
compute_loss=False,
ensemble_representations=True)
self.apply = jax.jit(hk.transform(_forward_fn).apply)
self.init = jax.jit(hk.transform(_forward_fn).init)
特征预处理
alphafold的推理其实就是调用RunModel的process_features()和predict()。
process_features的主要作用将raw feature dict进行tensor化和额外计算一些衍生的特征:

raw特征的处理细节由np_example_to_features模块的input_pipeline完成:
from alphafold.model.feature import np_example_to_features
features.np_example_to_features(
np_example=feature_dict,
config=model_config,
random_seed=random_seed)
主要步骤:
- make_data_config()根据模型的设置,确定需要后续处理的feature name, 比如用模板和不用模板分别需要处理的特征不同,可以在- cfg.common.unsupervised_features/cfg.common.template_features找到;
- 将FeatureDict中’deletion_matrix_int’换名’deletion_matrix’, 并且数据转为float32;
- np_to_tensor_dict()根据需要的根据feature_names创建空的tf的特征张量字典,并从raw_feature中将需要的数据拷走
- 关键步骤:input_pipeline.process_tensors_from_config中将可ensemble处理和非ensemble处理的feature单独处理,各类负责处理的data_transforms函数分别封装在nonensembled_map_fns()/ensembled_map_fns()中。 比如这里会对seq做bert masking,对msa做subsampling以及对template做residue mask等。
处理前的特征:
feature_dict.keys()
return: 返回的数据类型(**raw数据特征的预处理)
dict_keys(['aatype', 'between_segment_residues', 'domain_name', 'residue_index', 'seq_length', 'sequence', 'deletion_matrix_int', 'msa', 'num_alignments', 'msa_species_identifiers', 'template_aatype', 'template_all_atom_masks', 'template_all_atom_positions', 'template_domain_names', 'template_sequence', 'template_sum_probs'])
处理后的特征:
处理的细节可以参考alphafold原文–1.2.9 Featurization and model inputs
# setup using model_1:
model_name = 'model_1_ptm_pred_0'
model_runner = model_runners[model_name]
model_random_seed = 42
# processed_feature:
processed_feature_dict = model_runner.process_features(feature_dict, random_seed=model_random_seed)
processed_feature_dict.keys()
-----------------------------------------------------------------------
# return: 返回的数据类型(**数据特征的预处理)
dict_keys(['aatype', 'residue_index', 'seq_length', 'template_aatype', 'template_all_atom_masks', 'template_all_atom_positions', 'template_sum_probs', 'is_distillation', 'seq_mask', 'msa_mask', 'msa_row_mask', 'random_crop_to_size_seed', 'template_mask', 'template_pseudo_beta', 'template_pseudo_beta_mask', 'atom14_atom_exists', 'residx_atom14_to_atom37', 'residx_atom37_to_atom14', 'atom37_atom_exists', 'extra_msa', 'extra_msa_mask', 'extra_msa_row_mask', 'bert_mask', 'true_msa', 'extra_has_deletion', 'extra_deletion_value', 'msa_feat', 'target_feat'])
各种特征的含义与原文的对应关系(重点)
这里数据的维度E与recycling和Ensemble的设置有关。E= recycling+Ensemble,因此input的数据会有E个拷贝起始,但其中的数据并不是完全相同的,比如msa_feat和extra_msa_feat就是随机采样做sub sampling。【同时recycling因为jax需要jit的原因,所以拷贝的N份】
详细可查看alphafold补充材料:1.11.2 MSA resampling and ensembling章节
Separate batch per ensembling & recycling step.(input_pipeline)
N = MSA number. E = Number of ensemble+recycling. L = sequence length
Sequence&MSA特征:这里使用ensembling=0做展示
target_feat: shape = (E x L x 22) ,与补充材料不符,多了1维通道。代表target sequence的one-hot。
processed_feature_dict['target_feat'][0][0] # 索引第一个氨基酸
--------------------------------------------------------------------------
array([0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 1., 0.,
0., 0., 0., 0., 0.], dtype=float32)
aatype: : shape = (E x L),并不是原文中所述的one-hot representation,而是字母表list表示形式,这里限定为input sequence的序列。MSA的序列在true msa中记录。
processed_feature_dict['aatype'][0]
--------------------------------------------------------------------------
array([[14, 13, 0, 5, 9, 18, 10, 9, 6, 7, 1, 16, 6, 6, 5, 11,
1, 0, 19, 9, 6, 11, 19, 16, 5, 0, 12, 12, 6, 0, 19, 7,
0, 14, 11, 6, 2, 19, 1, 19, 17, 9, 8, 3, 19, 14, 11, 6,
2, 17, 7, 9, 7, 7, 19, 15, 0, 11, 0, 10, 7, 1]],
dtype=int32)
residue_index: shape = (E x L),input的序列编号,1维数据
processed_feature_dict['residue_index'][0]
--------------------------------------------------------------------------
array([[ 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15,
16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31,
32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47,
48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61]],
dtype=int32)
seq_length: shape = (E, ) input的序列长度,1维数据
processed_feature_dict['seq_length']
--------------------------------------------------------------------------
array([62], dtype=int32)
seq_mask: shape = (E x L), 全是1的矩阵,长度与input的序列长度相关,这里代表序列残基是否存在,存在=1,反之0(占位符)
processed_feature_dict['seq_mask']
--------------------------------------------------------------------------
array([[1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1.,
1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1.,
1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1.,
1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1.]],
dtype=float32)
msa_mask: shape = (E x 510 x L). 510可能是max MSA(每次这个数值貌似还会变),没有MSA序列比对的地方全是0,有msa序列的地方都是1. 这里的含义是,标记MSA矩阵中一共有多少条同源序列。(占位符)
processed_feature_dict['msa_mask']
--------------------------------------------------------------------------
array([[[1., 1., 1., ..., 1., 1., 1.],
[1., 1., 1., ..., 1., 1., 1.],
[0., 0., 0., ..., 0., 0., 0.],
...,
[0., 0., 0., ..., 0., 0., 0.],
[0., 0., 0., ..., 0., 0., 0.],
[0., 0., 0., ..., 0., 0., 0.]]], dtype=float32)
msa_row_mask: shape = (E x 510) 列版本的mask,那些列存在msa即标记为1,反之0。(占位符)
processed_feature_dict['msa_row_mask']
--------------------------------------------------------------------------
# 这里全是1说明,我把所有的msa通道都吃满了。
array([[1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1.,
1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1.,
1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1.,
1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1.,
...............................................................
1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1.,
1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1.,
1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1.,
1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1.]],
dtype=float32)
extra_msa: shape = (E x 5120 x L) , 在extra_msa中, 记录extra MSA序列的字母表list。
processed_feature_dict['extra_msa']
--------------------------------------------------------------------------
# extra MSA最大有5120条,这里我没有吃满。
array([[[14, 9, 9, ..., 10, 7, 11],
[14, 18, 19, ..., 10, 1, 1],
[14, 13, 0, ..., 10, 7, 1],
...,
[ 0, 0, 0, ..., 0, 0, 0],
[ 0, 0, 0, ..., 0, 0, 0],
[ 0, 0, 0, ..., 0, 0, 0]]], dtype=int32)
extra_msa_mask: shape = (E x 5210 x L) , 记录extra MSA序列是否存在的mask(占位符),注意第一条序列并不是input sequence。
processed_feature_dict['extra_msa_mask']
--------------------------------------------------------------------------
# extra MSA最大有5120条,这里我没有吃满。
array([[1., 1., 1., ..., 1., 1., 1.],
[1., 1., 1., ..., 1., 1., 1.],
[1., 1., 1., ..., 1., 1., 1.],
...,
[0., 0., 0., ..., 0., 0., 0.],
[0., 0., 0., ..., 0., 0., 0.],
[0., 0., 0., ..., 0., 0., 0.]], dtype=float32)
extra_msa_row_mask: shape = (E x 5210) , 列版本的extra MSA mask,那些列存在msa即标记为1,反之0。(占位符)
processed_feature_dict['extra_msa_row_mask']
--------------------------------------------------------------------------
array([[1., 1., 1., ..., 0., 0., 0.]], dtype=float32)
bert_mask: shape = (E x 510 x L),代表MSA中哪些位点被随机bert mask,mask的地方设置为1(占位符),反之0。每条序列被mask的地方其实都不一样。
processed_feature_dict['bert_mask'][0][0] # 索引第一条MSA序列的bert masking情况
--------------------------------------------------------------------------
array([0., 0., 0., 0., 0., 1., 0., 0., 0., 1., 0., 0., 1., 0., 0., 0., 1.,
1., 0., 0., 0., 0., 1., 1., 0., 1., 0., 0., 0., 1., 0., 1., 0., 0.,
1., 0., 0., 0., 0., 0., 0., 0., 1., 0., 0., 0., 0., 0., 0., 0., 0.,
0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 1.], dtype=float32)
true_msa: shape = (E x 510 x L),记录MSA序列的字母表list, 注意第一条序列即input sequence。
processed_feature_dict['true_msa'][0][0] # 索引第一条MSA序列的真实序列情况
--------------------------------------------------------------------------
array([14, 13, 0, 5, 9, 18, 12, 9, 6, 7, 1, 16, 6, 6, 5, 11, 11,
0, 19, 9, 6, 11, 19, 16, 0, 0, 10, 19, 21, 21, 21, 21, 21, 21,
21, 21, 2, 19, 1, 19, 17, 9, 8, 3, 19, 14, 11, 6, 2, 17, 7,
9, 0, 7, 5, 16, 0, 11, 6, 10, 7, 1], dtype=int32)
extra_has_deletion: shape = (E x 5120 x L), 指示extra MSAz中是否存在被随机crop删除的位点(占位符)。
processed_feature_dict['extra_has_deletion'][0][0] # 索引第一条MSA序列
--------------------------------------------------------------------------
array([0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0.,
0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0.,
0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0.,
0., 0., 0., 0., 0., 0., 0., 0., 0., 0., 0.], dtype=float32)
extra_deletion_value: shape = (E x 5120 x L), 指示MSA中被删除的氨基酸的占位符,被删除标记为1,反之0
processed_feature_dict['extra_deletion_value'][0]
--------------------------------------------------------------------------
array([[[0., 0., 0., ..., 0., 0., 0.],
[0., 0., 0., ..., 0., 0., 0.],
[0., 0., 0., ..., 0., 0., 0.],
...,
[0., 0., 0., ..., 0., 0., 0.],
[0., 0., 0., ..., 0., 0., 0.],
[0., 0., 0., ..., 0., 0., 0.]]], dtype=float32)
msa_feat: shape= (E x 510 x L x 49)***, constructed by concatenating “cluster_msa”, “cluster_has_deletion”, “cluster_deletion_value”, “cluster_deletion_mean”, “cluster_profile”.- cluster_msa: MSA cluster中心序列的one-hot representation, shape=(N x L x 23 ) (20 amino acids + unknown + gap + masked_msa_token).
- cluster_has_deletion: cluster中心序列是否存在deletion,shape = (N x L x 1)
- cluster_deletion_value: shape = (N x L x 1)
- cluster_deletion_mean: shape = (N x L x 1)
- cluster_profile: shape = (N x L x 1), cluster序列PSSM profile (one-hot), ,shape = (N x L x 23) (20 amino acids + unknown + gap + masked_msa_token).
注意看一下例子: 1-23 index代表cluster_msa的one-hot,27-49为PSSM的one-hot。
processed_feature_dict['msa_feat'][0][0][0] # 索引第一条MSA序列的第一号残基的融合特征。
--------------------------------------------------------------------------
array([0. , 0. , 0. , 0. , 0. ,
0. , 0. , 0. , 0. , 0. ,
0. , 0. , 0. , 0. , 1. ,
0. , 0. , 0. , 0. , 0. ,
0. , 0. , 0. , 0. , 0. ,
0. , 0. , 0. , 0. , 0. ,
0. , 0. , 0. , 0. , 0. ,
0. , 0. , 0. , 0. , 0.99999905,
0. , 0. , 0. , 0. , 0. ,
0. , 0. , 0. , 0. ], dtype=float32)
random_crop_to_size_seed: shape = (E x 2)
processed_feature_dict['random_crop_to_size_seed']
--------------------------------------------------------------------------
array([[-2104281239, -1605244653]], dtype=int32)
- Template的特征:这里使用ensembling=0做展示
N = top template number (default = 4). E = Number of ensemble+recycling. L = sequence length
template_aatype: shape = (E x N x L) 。代表的是模板的residue_id list。
processed_feature_dict['template_aatype'][0] # 这里我只用了top2模板,因此有2个。
--------------------------------------------------------------------------
array([[[14, 16, 10, 6, 19, 13, 10, 14, 0, 7, 8, 3, 3, 0, 1, 11,
0, 6, 10, 9, 0, 1, 10, 16, 7, 0, 16, 19, 3, 15, 9, 7,
0, 14, 9, 6, 15, 19, 1, 19, 10, 10, 16, 6, 10, 14, 0, 16,
8, 9, 7, 10, 7, 7, 1, 15, 0, 0, 3, 7, 0, 21],
[14, 9, 9, 5, 12, 2, 10, 10, 6, 7, 1, 16, 19, 6, 5, 11,
1, 2, 0, 19, 0, 0, 9, 16, 6, 0, 19, 19, 1, 16, 10, 3,
19, 1, 14, 3, 5, 19, 1, 9, 10, 9, 2, 6, 10, 7, 19, 6,
8, 13, 15, 19, 0, 7, 5, 16, 0, 0, 12, 1, 5, 21]]],
dtype=int32)
template_all_atom_masks:shape=(E x N x L x 37),以37维表示所有的原子占位符。表示L长度的序列,每个残基上都有哪些原子组成。atom_types可以在alphafold.commom.residue_constraint中找到。
processed_feature_dict['template_all_atom_masks'][0][0][0] # 索引模板1第一号氨基酸
--------------------------------------------------------------------------
array([1., 1., 1., 1., 1., 1., 0., 0., 0., 0., 0., 1., 0., 0., 0., 0., 0.,
0., 0., 0., 0., 0., 0., 0., 0.,
atom14字母表顺序:

template_all_atom_positions:shape=(E x N x L x 37 x 3),记录每个残基原子的xyz坐标,存在占位符的才有坐标。
processed_feature_dict['template_all_atom_positions'][0][0][0] # 索引模板1第一号氨基酸。
--------------------------------------------------------------------------
array([[-24.252, 35.212, -40.385],
[-24.195, 33.761, -40.554],
[-22.809, 33.228, -40.233],
[-24.539, 33.556, -42.033],
[-21.823, 33.908, -40.505],
[-24.406, 34.884, -42.665],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[-24.689, 35.894, -41.611],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ]], dtype=float32)
template_mask: shape = (E x N), 占位符=1,表示是否存在模板。
processed_feature_dict['template_mask'][0]
--------------------------------------------------------------------------
array([1., 1.], dtype=float32)
template_pseudo_beta: shape = (E x N x L x 3), pseudo_Cbeta的坐标,gap所在区域设置为(0,0,0)
processed_feature_dict['template_pseudo_beta'][0][0] # 索引第一个模板
--------------------------------------------------------------------------
array([[-24.539, 33.556, -42.033],
[-21.41 , 31.17 , -37.763],
[-19.807, 28.971, -42.991],
[-18.218, 24.974, -38.859],
[-15.743, 25.09 , -44.208],
...........................
[-10.062, 5.573, -42.15 ],
[-10.2 , 9.788, -45.781],
[ -6.04 , 7.021, -43.256],
[ -8.209, 2.633, -45.345],
[ -8.364, 5.342, -49.277],
[-12.928, 5.833, -47.695],
[ 0. , 0. , 0. ]], dtype=float32)
template_pseudo_beta_mask:shape = (E x N x L),pseudo_Cbeta的占位符,存在设置为1,反之0.
processed_feature_dict['template_pseudo_beta_mask'][0][0] # 索引第一个模板
--------------------------------------------------------------------------
array([1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1.,
1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1.,
1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 1.,
1., 1., 1., 1., 1., 1., 1., 1., 1., 1., 0.], dtype=float32)
atom14_atom_exists/atom37_atom_exists:shape = (E x L x 14/37) ,以atom14或atom37作为原子占位符的表示形式。这里的atom占位符指的是input sequence,而不是template。
processed_feature_dict['atom37_atom_exists'][0][61] # 索引第62位氨基酸
--------------------------------------------------------------------------
array([1., 1., 1., 1., 1., 1., 0., 0., 0., 0., 0., 1., 0., 0., 0., 0., 0.,
0., 0., 0., 0., 0., 0., 1., 0., 0., 0., 0., 0., 1., 1., 0., 1., 0.,
0., 0., 0.], dtype=float32)
atom14的定义:

residx_atom14_to_atom37: shape = (E x L x 14) 这里的含义是具体的原子号转换 ,这里的数值代表atom37的序号。
processed_feature_dict['residx_atom14_to_atom37'][0][61] # 索引第62位氨基酸
--------------------------------------------------------------------------
array([ 0, 1, 2, 4, 3, 5, 11, 23, 32, 29, 30, 0, 0, 0],
dtype=int32)
residx_atom37_to_atom14:shape = (E x L x 37) ,反之数值代表atom14的序号
processed_feature_dict['residx_atom37_to_atom14'][0][61] # 索引第62位氨基酸
--------------------------------------------------------------------------
array([ 0, 1, 2, 4, 3, 5, 0, 0, 0, 0, 0, 6, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 7, 0, 0, 0, 0, 0, 9, 10, 0, 8, 0,
0, 0, 0], dtype=int32)
PS:有一些文章中的特征比如以下,并没有在特征预处理这一步出现,可能是由于这并不是所有模型的通用输入。
- template_distogram
- template_torsion_angles/template_alt_torsion_angles
- template_torsion_angles_mask
- template_unit_vector
模型预设中,有部分的二面角特征是在alphafold模型的embeddings中进行处理的。
# The following models are fine-tuned from the corresponding models above
# with an additional predicted_aligned_error head that can produce
# predicted TM-score (pTM) and predicted aligned errors.
'model_1_ptm': {
'data.common.max_extra_msa': 5120,
'data.common.reduce_msa_clusters_by_max_templates': True,
'data.common.use_templates': True,
'model.embeddings_and_evoformer.template.embed_torsion_angles': True,
'model.embeddings_and_evoformer.template.enabled': True,
'model.heads.predicted_aligned_error.weight': 0.1
},
'model_2_ptm': {
'data.common.reduce_msa_clusters_by_max_templates': True,
'data.common.use_templates': True,
'model.embeddings_and_evoformer.template.embed_torsion_angles': True,
'model.embeddings_and_evoformer.template.enabled': True,
'model.heads.predicted_aligned_error.weight': 0.1
},
'model_3_ptm': {
'data.common.max_extra_msa': 5120,
'model.heads.predicted_aligned_error.weight': 0.1
},
'model_4_ptm': {
'data.common.max_extra_msa': 5120,
'model.heads.predicted_aligned_error.weight': 0.1
},
'model_5_ptm': {
'model.heads.predicted_aligned_error.weight': 0.1
}
模型推理
alphafoldv2目前需要输入model_random_seed,适配multimer。相对于monomer,ptm模型的输出多了pae(predicted_aligned_error), ptm相关的参数。
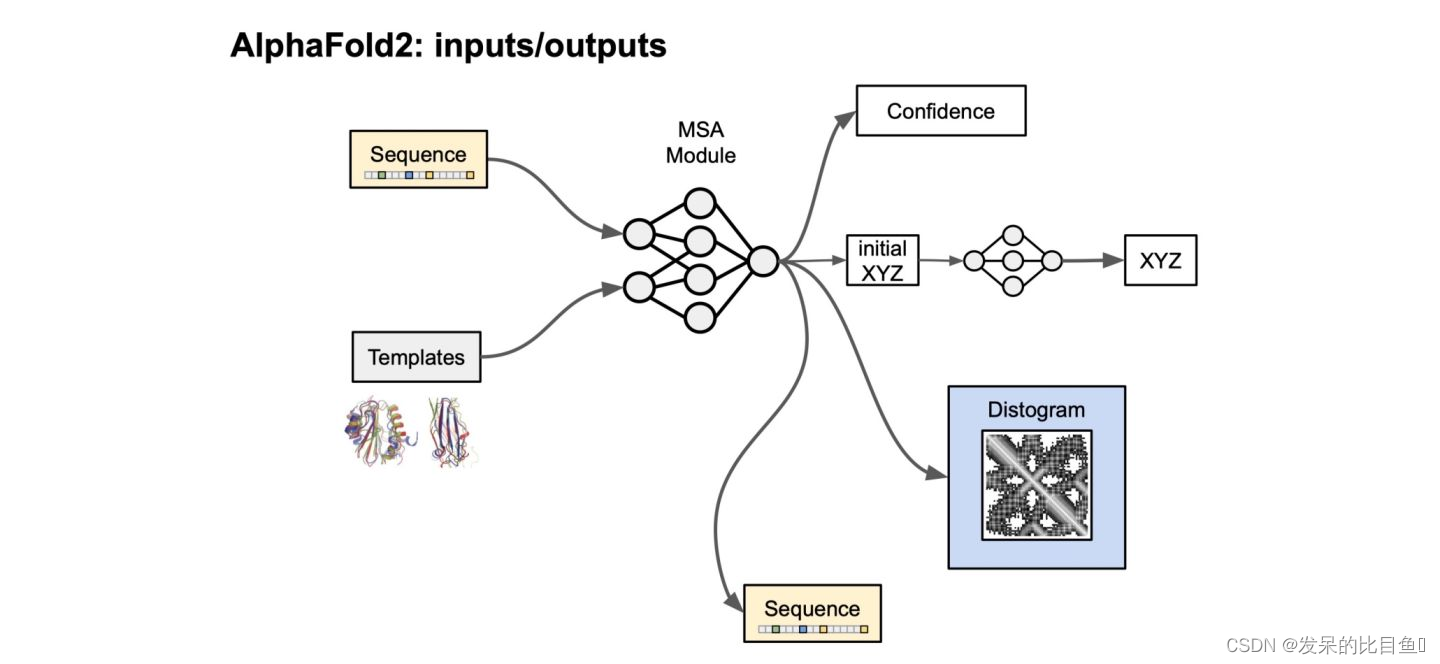
主要的api是model_runner.predict()函数:
prediction_result = model_runner.predict(processed_feature_dict,
random_seed=model_random_seed)
prediction_result.keys()
-----------------------------------------------------------------------
# return for model_0
dict_keys(['distogram', 'experimentally_resolved', 'masked_msa', 'predicted_lddt', 'structure_module', 'plddt', 'ranking_confidence'])
# return: model_ptm_0
dict_keys(['distogram', 'experimentally_resolved', 'masked_msa', 'predicted_aligned_error', 'predicted_lddt', 'structure_module', 'plddt', 'aligned_confidence_probs', 'max_predicted_aligned_error', 'ptm', 'ranking_confidence'])
一级封装:Class.RunModel()
如果解析更底层一些可以看看RunModel的类:
from alphafold.model import modules
class RunModel:
"""Container for JAX model."""
def __init__(self,
config: ml_collections.ConfigDict,
params: Optional[Mapping[str, Mapping[str, np.ndarray]]] = None):
def _forward_fn(batch):
model = modules.AlphaFold(self.config.model) # 实例化部分
return model(
batch,
is_training=False,
compute_loss=False,
ensemble_representations=True)
self.apply = jax.jit(hk.transform(_forward_fn).apply)
self.init = jax.jit(hk.transform(_forward_fn).init)
def process_features():
return x
def predict():
return y
实例化的部分的主干代码在alphafold.model.modules.AlphaFold()
batch: 指的是预处理得到的feature_dict;hk.transform将jax model转换为可以被即时编译的纯函数,得到init() 和 apply()
二级封装: Class.AlphaFold()
该类定义了外循环控制单次推理和recycling,步骤包括:
- 负责处理
msa的resampling和切feature batch - 输入
AlphaFoldIteration()进行推理获得recyle的prev字典

运行AlphaFold()主函数的注解解读:
from alphafold.model.modules import AlphaFold, AlphaFoldIteration
impl = AlphaFoldIteration(self.config, self.global_config)
# 定义get_prev func: 就是获取当前经过网络处理后的representation
def get_prev(ret):
new_prev = {
'prev_pos':
ret['structure_module']['final_atom_positions'],
'prev_msa_first_row': ret['representations']['msa_first_row'],
'prev_pair': ret['representations']['pair'],
}
return jax.tree_map(jax.lax.stop_gradient, new_prev)
# 定义do_call func:
def do_call(prev, recycle_idx, compute_loss=compute_loss):
if self.config.resample_msa_in_recycling:
num_ensemble = batch_size // (self.config.num_recycle + 1)
# 索引input的batch维,等于做resample_msa. 因为之前的resample的结果已经生成好了。维度与recycling number有关。
def slice_recycle_idx(x):
start = recycle_idx * num_ensemble
size = num_ensemble
# start即索引起始位点,size即向后切出size个数据;
return jax.lax.dynamic_slice_in_dim(x, start, size, axis=0)
ensembled_batch = jax.tree_map(slice_recycle_idx, batch)
else:
num_ensemble = batch_size
ensembled_batch = batch # ensembled的feature dict
non_ensembled_batch = jax.tree_map(lambda x: x, prev) # 载入prevs
# 关键是impl, 这里进行了一轮完整的AlphaFoldIteration.
return impl(
ensembled_batch=ensembled_batch,
non_ensembled_batch=non_ensembled_batch,
is_training=is_training,
compute_loss=compute_loss,
ensemble_representations=ensemble_representations)
# 初始化创建一个prev_representation的dict;
prev = {}
# prev xyz
prev['prev_pos'] = jnp.zeros([num_residues, residue_constants.atom_type_num, 3])
# prev row1 msa feature
prev['prev_msa_first_row'] = jnp.zeros([num_residues, emb_config.msa_channel])
# prev pair feature
prev['prev_pair'] = jnp.zeros([num_residues, num_residues, emb_config.pair_channel])
# 定义了一个匿名函数来控制循环,跑get_prev和do_call:
body = lambda x: (x[0] + 1, # pylint: disable=g-long-lambda
get_prev(do_call(x[1], recycle_idx=x[0],
compute_loss=False)))
# 实际运行n-1 recycling的操作
if hk.running_init():
# When initializing the Haiku module, run one iteration of the
# while_loop to initialize the Haiku modules used in `body`.
_, prev = body((0, prev))
else:
_, prev = hk.while_loop(
lambda x: x[0] < num_iter,
body,
(0, prev))
# 最后一轮:
ret = do_call(prev=prev, recycle_idx=num_iter)
if not return_representations:
del (ret[0] if compute_loss else ret)['representations'] # pytype: disable=unsupported-operands
return ret #<--- outputs here;
三级封装:Class.AlphaFoldIteration()
这里负责处理单次推理与repr ensemble的处理,核心部件是EmbeddingsAndEvoformer和Head_Factory。
基本处理过程:
- 执行
EmbeddingsAndEvoformer和ensemble化其部分output; - 创建
head_factory初始化各类head的hk module; - 执行各种
heads,各类loss的计算,返回result dict。

特别值得注意的是:
msa repr并没有进行ensemble处理,而是直接取batch0的结果,其他的repr均进行平均化处理;head_factory负责处理各类的head,其中也包括了structure module。
注:所有的alphafold原文补充材料中的算法都在这个模块中实现。(这里不做赘述,神经网络的算法代码对着文章很好读)
模型输出
重要的输出数据类型解释参考官方github解释
distogram: 2D_features, logits: NumPy array of shape [N_res, N_res, N_bins]. N_bins = 64。将contact map距离分为了64个bin,每个bin含有的是分布概率。ranking_confidence: 模型的打分排名,用于最后模型排序:
prediction_result['ranking_confidence'] = 84.29389819306218
Structure Embeddings: 模型输出的结构信息可以在此找到,与raw feature特征直接相关:
prediction_result['structure_module']
--------------------------------------------------------------
dict_keys(['final_atom_mask', 'final_atom_positions'])
final_atom_mask/final_atom_positions: 原子坐标 37维,对应不同元素的xyz坐标
prediction_result['structure_module']['final_atom_mask'].shape = (seq_len, 37)
prediction_result['structure_module']['final_atom_positions'].shape = (seq_len, 37, 3)
output as pdbfile: 将embeddings转换为pdb 人类可读的3D坐标信息:
from alphafold.common import protein
from alphafold.common import residue_constants
# output as PDB files:
# Add the predicted LDDT in the b-factor column.
# Note that higher predicted LDDT value means higher model confidence.
plddt = prediction_result['plddt']
plddt_b_factors = np.repeat(plddt[:, None], residue_constants.atom_type_num, axis=-1)
unrelaxed_protein = protein.from_prediction(
features=processed_feature_dict,
result=prediction_result,
b_factors=plddt_b_factors,
remove_leading_feature_dimension=not model_runner.multimer_mode)
pdb_strings = protein.to_pdb(unrelaxed_protein)
plddt: 每个residue残基的pLDDT打分,维度为L,数值范围0-100,越高代表残基结构的置信度越高。
array([86.63831682, 89.79428545, 87.84879985, 89.85212798, 88.03631921,
86.12329863, 86.40511123, 84.75732675, 85.99181068, 88.14490297,
86.83076695, 91.81795432, 91.07235044, 93.74915771, 90.97843239,
91.48212398, 92.79137088, 93.86457783, 92.1685246 , 92.63566183,
94.01498471, 94.59793215, 92.62430178, 93.71278408, 93.65539971,
92.75957977, 90.83720729, 92.91265442, 94.08131697, 91.84352532,
89.68613155, 89.96687109, 87.03115079, 90.97443059, 89.12853574,
92.0847665 , 91.49042418, 91.71996175, 92.59472258, 91.44372126,
92.14367958, 89.89447384, 88.85395624, 88.55568358, 85.36568714,
83.92866688, 81.00400393, 77.495124 , 74.78476173, 74.71061615,
66.47940765, 65.12609518, 60.34433093, 59.79908405, 65.70828864,
65.76185942, 65.37726497, 66.25180978, 64.66977383, 63.97790086,
64.30624492, 53.53935194])
ptm: predicted TM-score. 标量,评估全局的superposition metric。这个指标的代表全局结构的packing质量评估。predicted_aligned_error: 维度为LxL,数值范围为0-max_predicted_aligned_error。0代表最可信,该指标也可以作为domain packing质量的评估。
来源
https://zhuanlan.zhihu.com/p/492381344

瓜分20万奖金 获得内推名额 丰厚实物奖励 易参与易上手
更多推荐

 4
4 0
0- 0



 已为社区贡献3条内容
已为社区贡献3条内容

所有评论(0)