
论文解读:《多层肽 - 蛋白质相互作用预测的深度学习框架》
期刊naturecommunications。影像因子17.694。
Title:A deep-learning framework for multi-level peptide–protein interaction prediction
期刊:nature communications
分区:1区
影像因子:17.694
代码数据集:GitHub - twopin/CAMP: predicting peptide-protein interactions
目录
1. 摘要
肽 - 蛋白质相互作用参与了各种基本细胞功能,它们的鉴定对于设计有效的肽疗法至关重要。最近,已经开发了许多计算方法来预测肽 - 蛋白质相互作用。但是,大多数现有的预测方法在很大程度上取决于高分辨率结构数据。在这里,我们提出了一个深度学习框架,用于多级肽蛋白相互作用预测,称为CAMP,包括二进制肽 - 蛋白质相互作用预测和相应的肽结合残基鉴定。全面的评估表明,CAMP可以成功捕获肽和蛋白质之间的二元相互作用,并识别沿相互作用所涉及的肽的结合残基。此外,CAMP在二元肽蛋白相互作用预测方面的表现优于其他最先进的方法。 CAMP可以用作肽蛋白相互作用预测和鉴定肽中重要结合残基的有用工具,从而可以促进肽药物发现过程。
2. 数据集
构建了一个基准数据集,即来自RCSB PDB21,22的蛋白质 - 肽复合物结构,以及来自Drugbank的已知药物对接对(可以在补充注释10和相应的PDB ID中找到更多数据策划的详细信息我们用于培训和测试的方法可以在补充数据的补充表12和13中找到。我们使用的药品银行ID可以在补充数据中的补充表14中找到)。总体而言,我们获得了7417个阳性相互作用对,涵盖了3412个蛋白序列和5399个肽序列。其中,来自RCSB PDB的6581对具有肽序列中的残基级结合标签。然后,我们通过随机将那些非相互作用的蛋白质和肽对构建了负数据。更具体地说,对于每种正相互作用,通过从所有改组对的非相互作用蛋白和肽对随机采样来产生五个负面因素。总体而言,作为基准数据集,我们获得了44,502个肽 - 蛋白质对。
判断肽序列长度是否有50,小于50的要进行填充
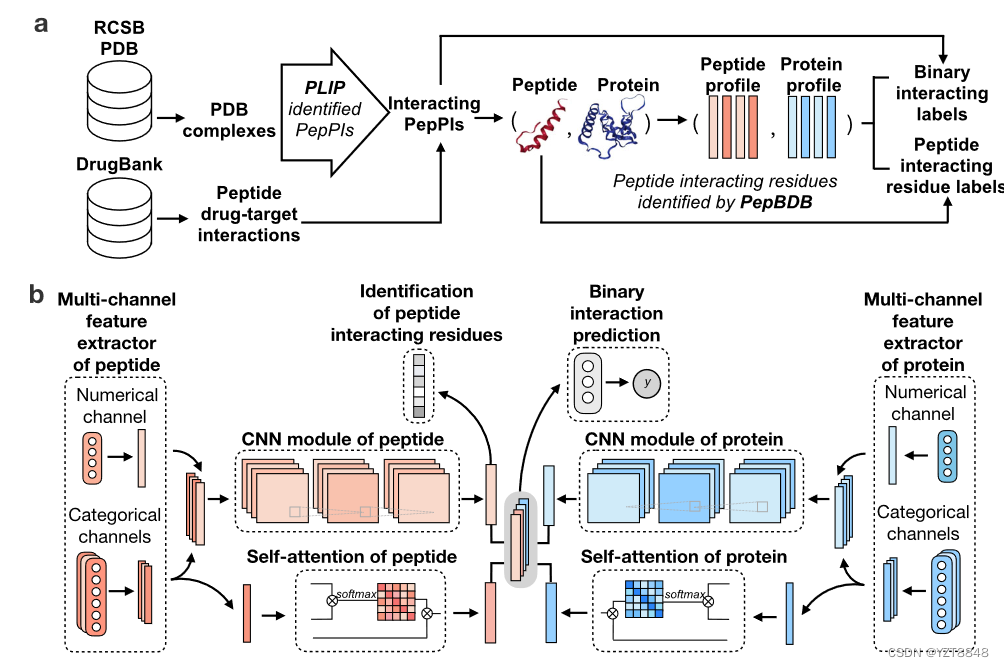
3. 方法
为了避免轮廓内不同特征的不一致尺度(即,疾病和PSSM特征是密集的矢量,而残留级的特性是分类的矢量),CAMP利用了两个多通道特征提取器来得出编码的特征,这些特征是处理蛋白质和肽的处理。分别剖面。每个提取器都有三个分类通道和一个数值通道(图1B)。每个分类通道都由三个自学习单词嵌入层30组成,分别以氨基酸,二级结构和生理化学表示为输入。每个数值通道都由一个完全连接的层组成,以将密集特征作为输入,即肽和蛋白质的内在障碍趋势特征(范围为0至1之间)以及蛋白质的归一化进化基质(PSSM)。
在这里,设计了这样的多通道体系结构,因为输入配置文件包含不同尺度的多方面特征,如果我们只使用简单的编码器,它们可能会带来不一致的情况。接下来,CAMP利用了两个卷积神经网络(CNN)模块,它们分别提取肽和蛋白质的隐藏上下文特征。此外,CAMP采用自我注意的机制来学习残基与蛋白质和肽对最终相互作用预测的单个残基之间的长期依赖性。之后,Camp结合了所有提取的特征,并使用三个完全连接的层来预测给定的相互作用。
4. 结果
图2还显示,在某些集群设置下,CAMP生成了相对变化的预测结果。为了进一步研究引起这种现象的潜在因素,我们使用二进制预测任务的五倍交叉验证程序进行了其他分析(在补充注释1中)。我们的分析结果(补充图1)表明,在两个聚类设置下的相对较大的预测误差可能是由某些蛋白质家族,结构域和生物(例如,用于蛋白质家族的组蛋白和GPCR,用于域的组蛋白和GPCR),以及用于结构域的胰蛋白酶和kringle,以及蛋白质生物的牛)。此外,我们进行了全面的消融研究,以证明营地各个组成部分的重要性,包括不同的特征组和网络体系结构中的自我发项模块(补充注释2)。我们的消融研究(补充表2和补充图2)表明,当前的模型体系结构和特征选择方案对于我们的预测任务是最佳的。通过表征肽上的结合残基来表征新见解。到目前为止,已经开发了许多计算方法来预测PEPPI预测中蛋白质表面上的相互作用位点14,34,35。这些方法从肽 - 蛋白质复合物的3D结构信息中学习,并可以以相对良好的精度在蛋白质表面上查明相互作用的位点。然而,很少有模型专门设计用于表征辣椒肽上的相互作用位点,这对于理解肽的生物学作用和设计有效的肽药物也至关重要。对于药理学家而言,Thechoiceoficalmicalmification在很大程度上依赖于鉴定与结合活性有关的必需肽残基1。从传统上讲,药理学家会迭代地替换可能的残基并进行湿法进行验证。尽管这些尝试可以为进一步的药物设计提供有用的信息,例如,在其侧链上改变特定的非结合残基或修改组以提高稳定性并降低毒性1,2,但这些实验方法通常昂贵且耗时。在CAMP中,我们设计了一个监督预测模块,以鉴定肽序列中的结合残基。我们首先使用源自PEPBDB29的相互作用信息构建了一组合格的标签,用于肽结合残基,该信息是一个综合结构数据库,其中包含来自RCSB PDB21,22的已知相互作用肽 - 蛋白质复合物
5.结论
在这项工作中,我们提出了camp,这是一个多级肽 - 蛋白质相互作用预测的深度学习框架,包括二元相互作用预测和肽结合残基预测。我们首先生成了一系列基于序列的特征,以构建用于肽和蛋白质的特征曲线。与传统的肽或蛋白质特征表示(例如K-MER)相比,我们的综合特征曲线结合了内容丰富的结构注释特征,进化信息和内在障碍趋势得分,以增强肽 - 蛋白质相互作用的预测。然后,我们使用多通道功能提取器分别处理数值和分类特征,以避免多源特征的不一致。全面的交叉验证评估表明,cAMP在二进制相互作用预测上的最先进基线方法的表现优越。此外,我们试图通过鉴定肽结合残基来破译肽 - 蛋白质相互作用的潜在机制。我们表明cAMP可以准确检测到肽序列中的结合残基。我们还提出了四个代表性的案例,以可视化肽结合残基识别任务的结果,并检查了Semaglutide及其类似物的预测目标。我们还验证了CAMP在肽-PBD相互作用预测中的应用潜力,肽 - 蛋白质对的结合亲和力评估以及肽的虚拟筛查。所有这些结果表明,CAMP可以提供准确的肽 - 蛋白质相互作用预测以及对理解肽结合机制的有用见解。与基于结构的对接方法相比,CAMP提供了各种优势。例如,CAMP可以同时完成预测二进制相互作用并识别相互作用中涉及的肽结合残基的任务,而先前基于结构的方法仅着眼于预测结合姿势或识别蛋白质表面上的结合区域。此外,对于单个肽 - 蛋白质对,CAMP在几秒钟内进行预测,而基于结构的对接方法通常需要几个小时。此外,CAMP仅需要序列信息作为输入,因此不依赖有限的结构数据。更具体地说,在瑞士prot数据库中有564,638个蛋白质具有手动注释序列信息,但只有8.49%的蛋白质具有已解决的结构。在这种情况下,CAMP能够比当前基于结构的方法对靶蛋白进行更多的靶蛋白进行预测,从而具有更广泛的应用。然而,当前版本的CAMP仍然存在某些局限性。例如,它不能直接预测给定肽 - 蛋白质对中蛋白质序列的结合残基。实际上,我们探索了CAMP是否可以预测蛋白质的结合残基。在五倍交叉验证的“随机设置”下,当添加一个预测蛋白质结合残基的模块时,CAMP确定了少于实际结合残基的20%不到20%,而二进制交互预测任务的平均AUC略微降低到0.843。在我们框架中,对蛋白质结合残留预测的相对不满意的结果可能是由于以下挑战所致。首先,蛋白质序列通常比肽长得多,范围从52至4911残基,在固定确切的相互作用残基方面构成了困难。其次,使用PLIP从共晶体复合物结构中提取蛋白质结合残基的阳性标记时可能会出现某些不确定性。将来,我们计划合并更多数据,例如结合域信息,以进一步改善预测蛋白质中结合残基的结果。
看不太懂,以后尽量避免这类文章

更多推荐

 2
2 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)