
手把手教学差异表达基因分析
文章目录引言安装并导入DESeq2包数据要求制作dds对象,进行差异分析筛选差异基因完整代码引言对于组学分析来说,常常会寻找组间的差异,例如差异基因(转录组)、差异菌(宏基因组)以及差异通路(宏基因组),而转录组分析上最为经典的DESeq2包对于以上分析也都适用DESeq最早在2010年发表在Genome Biology上,2014年上更新版本DESeq2。DESeq2是基于负二项广义线性模型估算
引言
对于组学分析来说,常常会寻找组间的差异,例如差异基因(转录组)、差异菌(宏基因组)以及差异通路(宏基因组),而转录组分析上最为经典的DESeq2包对于以上分析也都适用
DESeq最早在2010年发表在Genome Biology上,2014年上更新版本DESeq2。DESeq2是基于负二项广义线性模型估算样本间基因差异表达概率,既适于有生物学重复的也适于没有生物学重复的样本,同时除了在转录组学,在宏基因组上使用DESeq2计算差异菌的也有报道

安装并导入DESeq2包
DESeq2包目前需要依赖BiocManager包,该包用于管理bioconductor项目中的所有包,因此首先安装该包
# 1.判断是否有BiocManager包,若不存在则安装
options(repos=structure(c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))) #设置清华镜像,加速下载
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
if (!requireNamespace("DESeq2", quietly = TRUE))
BiocManager::install('DESeq2') #通过BiocManager安装DESeq2
library(DESeq2) #加载library
由于DESeq2包较大,安装时间较长,当导入后没有任何报错即为安装成功
数据要求
- 1.基因表达矩阵
行为基因,列为样本,需要注意表达数据为原始reads count,DESeq2内部会根据样本大小对counts进行调整,自带标准化过程,不要使用标准化后的FPKM或TPM

- 2.样本分组信息
样品信息,第一列是样品名称,第二列是样品的处理情况(对照还是处理等),即condition,condition的类型是一个factor,第三列可添加其他信息(可选)。需要自己根据临床/实验分组信息单独建立

制作dds对象,进行差异分析
想要进行差异分析,首先需要生成DESeq2必须的数据类型,即dds类型数据,DESeq2包使用DESeqDataSet(dds)作为存放count数据及分析结果的对象,每个DESeqDataSet(dds)对象都必须由以下三者:
- countData,存放counts matrix的对象
- colData,存放分组信息和处理信息的对象
- design公式,对应sample的分组信息,需要以~ 波浪字符进行连接,而不同的信息之间需要以+连接,示例:
design=~variable_1 + variable_2
这些分组信息会被用来估算离散度和估算Log2 fold change的模型
注意,为了方便起见,在默认参数下,用户应该把感兴趣的分组信息放在formula的最后,并且确认control组的level是第一个(因子的level,见R语言基础视频)
## 制作dds对象,构建差异基因分析所需的数据格式
dds <- DESeqDataSetFromMatrix(countData = B, colData = coldata, design = ~ condition);
# countData = B,readscount矩阵
# colData = coldata,分组信息,根据这个才能在两组之间比较
# design = ~ condition,公式,表示按照condition进行分析
## 5.差异分析结果
dds <- DESeq(dds) #正式进行差异分析
dds对象包含的内容:

详细差异分析步骤:
estimating size factors:估算库size factor
estimating dispersions:估算离散度
gene-wise dispersion estimates:估算基因的离散度
mean-dispersion relationship:均值-离散度关系
final dispersion estimates:估算最终离散度
fitting model and testing:模型适应和测试
差异分析完成后,使用results从DESeq分析中提取出一个结果表,从而给出样品的基本均值,log2倍变化,标准误差,测试统计量,p值和校整后的p值

筛选差异基因
一般按照pvalue < 0.05 & |log2FoldChange| >1筛选差异基因,如果想要结果更可靠,可选择使用更严谨的padj和更大的log2FoldChange进行筛选
最后可分别保存差异上调和差异下调的基因:
filter_up <- subset(res, pvalue < 0.05 & log2FoldChange > 1) #过滤上调基因
filter_down <- subset(res, pvalue < 0.05 & log2FoldChange < -1) #过滤下调基因
得到差异基因之后,可进行基因功能富集、PCA和热图聚类等后续分析
完整代码
# <差异基因分析>
# 1.判断是否有BiocManager包,若不存在则安装
options(repos=structure(c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))) #设置清华镜像,加速下载
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
if (!requireNamespace("DESeq2", quietly = TRUE))
BiocManager::install('DESeq2') #通过BiocManager安装DESeq2
library(DESeq2) #加载library
setwd("C:/Users/yut/Desktop/data") #设置工作目录,所有输出文件保存于此
#输入数据要求
# DEseq2要求输入数据是由整数组成的矩阵
# DESeq2要求矩阵是没有标准化的
##2.读入所有基因原始readscount表达矩阵,行为基因,列为样品
A <- read.table("control_case_readscount.txt", header = T, row.names = 1)
B <- as.matrix(A) #转换成矩阵格式,保证都是数值
View(B)
## 3.实验分组
# 样品信息矩阵即上述代码中的colData,它的类型是一个dataframe(数据框),
# 第一列是样品名称,第二列是样品的处理情况(对照还是处理等),即condition
coldata <- read.table("sample_info.txt",header = T,row.names = 1)
coldata <- coldata[, c("condition", "type")]
View(coldata) #查看分组信息
## 4.制作dds对象,构建差异基因分析所需的数据格式
dds <- DESeqDataSetFromMatrix(countData = B, colData = coldata, design = ~ condition);
# countData = B,readscount矩阵
# colData = coldata,分组信息,根据这个才能在2组之间比较
# design = ~ condition,公式,表示按照condition进行分析
## 5.差异分析结果
dds <- DESeq(dds) #正式进行差异分析
## 6.提取结果,在treated和untreated组进行比较
res <- results(dds, contrast = c("condition", "treated", "untreated"))
# results从DESeq分析中提取出一个结果表,从而给出样品的基本均值,log2倍变化,标准误差,测试统计量,p值和校整后的p值;
resordered <- res[order(res$padj),] #按照padj从小到大排序
sum(res$padj < 0.05, na.rm = TRUE) #统计padj小于0.05显著差异的基因
## 7.输出图片
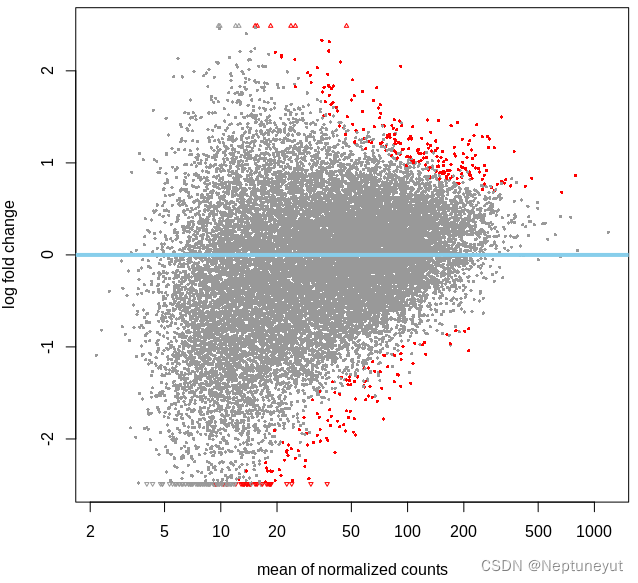
plotMA(res) #画火山图,横轴是标准化后的平均readscount,纵轴是差异倍数,大于0是上调,小于0是下调,红色点表示显著差异的基因
plotMA(res, alpha = 0.05, colSig = 'red', colLine = 'skyblue')
# alpha:p-value
# colSig:显著性基因的颜色
# colLine:y=0的水平线
##8.过滤上调、下调基因
filter_up <- subset(res, pvalue < 0.05 & log2FoldChange > 1) #过滤上调基因
filter_down <- subset(res, pvalue < 0.05 & log2FoldChange < -1) #过滤下调基因
print(paste('差异上调基因数量: ', nrow(filter_up))) #打印上调基因数量
print(paste('差异下调基因数量: ', nrow(filter_down))) #打印下调基因数量
##9.保存到文件
write.table(as.data.frame(resordered), file = "./differential_gene.txt") #log2FoldChange + pvalue + padj
write.table(filter_up, file="./filter_up_gene.txt", quote = F)
write.table(filter_up, file="./filter_down_gene.txt", quote = F)
- MA-plot (M-versus-A plot)
MA图对数据分布情况的可视化。该图将数据转换为M(对数比)和 A(平均值),然后绘制这些值来可视化两个样本中测量值之间的差异。
其他问题
- Error in checkSlotAssignment
> ## 6.提取结果,在treated和untreated组进行比较
> res <- results(dds, contrast = c("condition", "treated", "untreated"))
Error in checkSlotAssignment(object, name, value) :
assignment of an object of class “DFrame” is not valid for slot ‘elementMetadata’ in an object of class “DESeqResults”; is(value, "DataTable_OR_NULL") is not TRUE
解决方法:
尝试使用.libPaths()查看DESeq2的位置,并找到删除,再重新运行安装命令,Update选择all.

旨在为数千万中国开发者提供一个无缝且高效的云端环境,以支持学习、使用和贡献开源项目。
更多推荐

 44
44 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)