




- @qq_20563997
简介
该用户还未填写简介
擅长的技术栈
可提供的服务
暂无可提供的服务
virtualbox7.0以上版本,如果想改变一下安装目录,比如安装D盘,会出现报错无法安装。官方给出了相应的解决方案:https://www.virtualbox.org/manual/ch02.html#install-win-installdir-req。

主要分成三步:一、结构优化;二、静态自洽计算;三、非自洽计算以Al-FCC为例子。
载流子迁移率通常指半导体内部电子和空穴整体的运动快慢情况,是衡量半导体器件性能的重要物理量。2004年,石墨烯的成功剥离引起了研究人员对于二维材料性质探索的浓厚兴趣。石墨烯、黑磷等二维材料展现出的高载流子迁移率是其中的一个重要研究课题,科研人员在理论计算方面已经做了大量的工作。由于电子在运动过程中不仅受到外电场力的作用,还会不断的与晶格、杂质、缺陷等发生无规则的碰撞,大大增加了理论计算的难度。
摘要:本文通过VASP软件计算氢原子能量,比较了不同计算方法的结果。采用LSDA近似时,氢原子能量为-12.92eV;引入自旋限制(ISPIN=2)后,能量提高约0.88eV;改用GGA赝势后,计算结果更接近理论值-13.6eV,达到-13.67eV。文章还讨论了计算参数的设置,包括单K点采样、INCAR文件配置等,并指出由于使用赝势,对所得能级的解释存在困难,欢迎讨论。
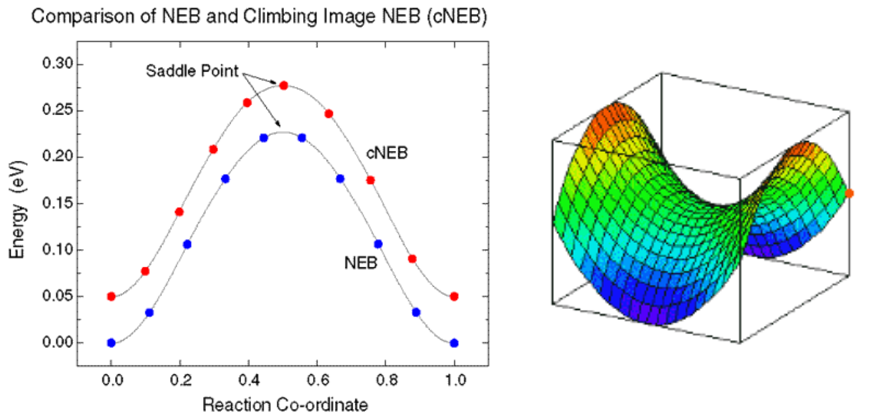
使用VASP计算过渡态,最重要的步骤就是在得到了初态和末态结构之后,进行反应路径的插值,插值的优劣直接关系到后面的过渡态搜索的难易。
关于VASP计算不收敛的解决方法,有些帖子已经总结的很好了,可以根据自己遇到的情况直接拿来用。如果把这些方法都用了还是不行,恐怕是你的结构本身问题较大了。1. 帖子1:解决VASP计算SCF/几何优化/过渡态不收敛的方法总结2. 帖子2:关于VASP计算不收敛的一些实例与解决办法3. 官方:Difficult to converge systems。
