单细胞分析(五)——使用Harmony进行数据整合和去批次
为了保证单细胞RNA测序数据的准确性和可靠性,需要对数据进行样本去批次处理。去批次的方法包括Harmony,SeuratV3等,这些方法可以有效地消除批次效应和技术噪声,并提高单细胞RNA测序数据的质量,为后续的数据分析提供更加可靠的基础。
为什么要进行数据去批次
进行样本去批次(batch correction)是单细胞RNA测序数据分析的重要步骤之一。
技术噪声和批次效应: 单细胞RNA测序数据通常具有高度异质性,且在采样、实验操作、反应条件等多个环节中可能引入技术噪声和批次效应。这些因素会对测序数据质量产生影响,从而使得不同批次数据之间存在较大的差异。
批次效应的干扰:不同批次数据之间的差异会掩盖真正的生物学差异,从而影响后续的分析结果。例如,在聚类分析中,如果没有进行批次校正,就很容易把来自不同批次的细胞划分到不同的亚群中。
提高可比性和统计功效:去除批次效应可以提高不同实验之间的数据可比性,也可以增加数据之间的统计功效,从而更好地发现生物学上的差异和特点。
因此,为了保证单细胞RNA测序数据的准确性和可靠性,需要对数据进行样本去批次处理。去批次的方法包括Harmony,SeuratV3等,这些方法可以有效地消除批次效应和技术噪声,并提高单细胞RNA测序数据的质量,为后续的数据分析提供更加可靠的基础。
数据分次读入
使用GSE135337这个数据集中的数据进行分析
将数据处理成seurat对象 格式,然后保存后,进行分析比较,直接读入,进行分析。
data1 <- readRDS('./output_data/GSE135337_scobj/BC1.RDS')
data2 <- readRDS('./output_data/GSE135337_scobj/BC2.RDS')
data3 <- readRDS('./output_data/GSE135337_scobj/BC3.RDS')
data4 <- readRDS('./output_data/GSE135337_scobj/BC4.RDS')
data5 <- readRDS('./output_data/GSE135337_scobj/BC5.RDS')
data6 <- readRDS('./output_data/GSE135337_scobj/BC6.RDS')
datan <- readRDS('./output_data/GSE135337_scobj/BCN.RDS')
### 把数据变成list
data <- list(data1, data3, data4, data5, data6, datan)
多个数据整合
Merge 合并数据
### Merge 合并数据
scobj <- merge(x=data[[1]], y = data[-1], project = "scBC")
## 删除所有data前缀的数据,释放空间
rm(list = ls(pattern="data.*"))
数据去批次处理
在去批次处理之前,需要主成分分析降维后的数据,因此还需要把这一部分数据进行统一降维
数据质控
质控过程中seurat对象保存的metadata中,nFeature_RNA, number of Feature, 每个细胞中有多少个基因,nCount_RNA, number of counts, 每个细胞中有多少个counts,percent.mt, 自己增加的列, 每个细胞中线粒体基因的比例
### 3.数据质控
scobj[["percent.mt"]] <- PercentageFeatureSet(scobj, pattern = "^MT-")
#上面的函数进行的操作 scobj@meta.data$percent.mt <- PercentageFeatureSet(scobj, pattern = "^MT-")
### 该操作会在metadata数据里面增加一列叫做percent.mt
### 质控数据可视化,使用VlnPlot函数
VlnPlot(scobj, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
### 正式筛选,筛选的是细胞,最终细胞减少
# 根据具体情况进行筛选阈值的确定
scobj <- subset(scobj, subset = nFeature_RNA > 500 & nFeature_RNA < 6000 & percent.mt < 10)
scobj <- NormalizeData(scobj)
scobj <- FindVariableFeatures(scobj, selection.method = "vst", nfeatures = 2000)
scobj <- ScaleData(scobj, features = rownames(scobj))
scobj <- RunPCA(scobj, features = VariableFeatures(object = scobj),reduction.name = "pca")
ElbowPlot(scobj)
DimPlot(scobj, reduction = "pca")
### 不进行批次矫正
scobj <- RunUMAP(scobj,reduction = "pca", dims = 1:10, reduction.name = "umap_naive")
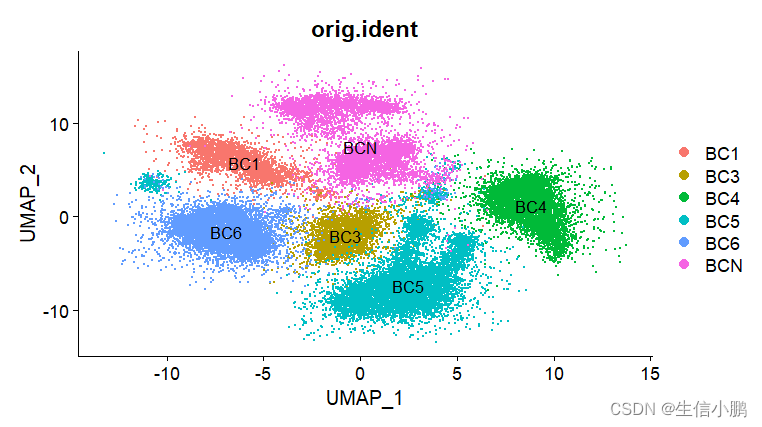
p1 <- DimPlot(scobj, reduction = "umap_naive",group.by = "orig.ident")
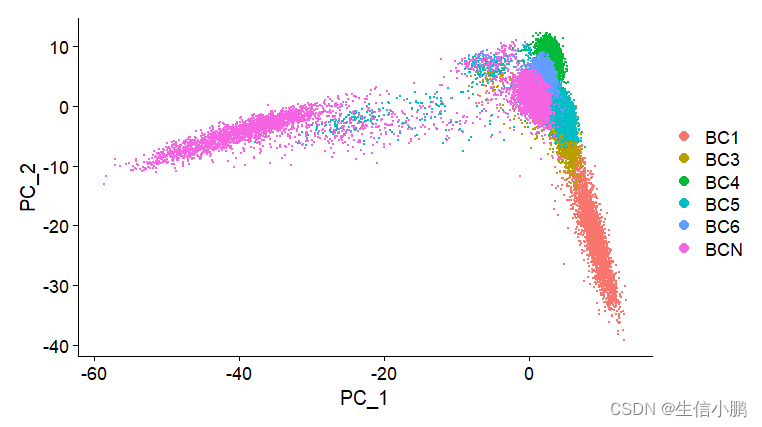
这里降维分析过程中,RunUMAP函数输入的是PCA数据,输出是在reduction中按照所命名的名字进行,也就是reduction.name这个参数
这里再将展示主成分解释方差的内容进行具体化
## 查看数据的复杂程度
ElbowPlot(scobj, reduction = "pca", ndims = 30)
xx <- cumsum(scobj[["pca"]]@stdev^2)
xx <- xx / max(xx)
which(xx > 0.9) # 可以查看多少PCs解释了90%的方差,假设10%的方差来自于噪声,然后就可以选择相应的PCs
ndim = 33 # dims的参数
去批次后数据分析
library(harmony)
scobj <- RunHarmony(scobj,reduction = "pca",group.by.vars = "orig.ident",reduction.save = "harmony")
scobj <- RunUMAP(scobj, reduction = "harmony", dims = 1:30,reduction.name = "umap")
把去批次后的进行样本降维展示
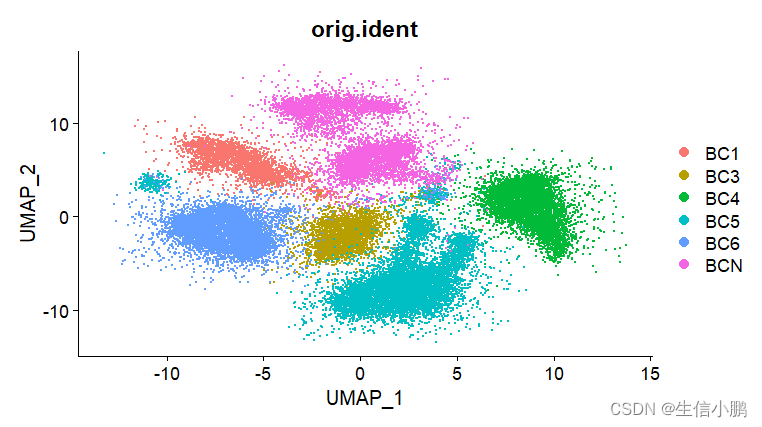
p2 <- DimPlot(scobj, reduction = "umap",group.by = "orig.ident", label = T)
可以看出去批次之前,研究项目之间的批次效应是比较明显的,即便是类似的疾病样本,其分开的还是十分明显,去批次后,虽然图看起来没有那么漂亮,但是其说明相似的细胞是聚在一起的。
后续分析
scobj <- FindNeighbors(scobj, reduction = "harmony", dims = 1:30)
### 设置多个resolution选择合适的resolution
scobj <- FindClusters(scobj, resolution = seq(0.2,1.2,0.1))
参考文章
Integration of datasets using Harmony
https://github.com/immunogenomics/harmony

为开发者提供学习成长、分享交流、生态实践、资源工具等服务,帮助开发者快速成长。
更多推荐



 6
6 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)