

SRA文件的下载(prefetch)和解压SRA文件(fastq-dump)
sra文件下载方式NCBI-SRA和EBI-ENA数据库SRA数据库: Sequence Read Archive:隶属NCBI (National Center for Biotechnology Information),它是一个保存大规模平行测序原始数据以及比对信息和元数据 (metadata) 的数据库,所有已发表的文献中高通量测序数据基本都上传至此,方便其他研究者下载及再研究。其中的数据
sra文件下载方式
NCBI-SRA和EBI-ENA数据库
SRA数据库: Sequence Read Archive:隶属NCBI (National Center for Biotechnology Information),它是一个保存大规模平行测序原始数据以及比对信息和元数据 (metadata) 的数据库,所有已发表的文献中高通量测序数据基本都上传至此,方便其他研究者下载及再研究。其中的数据则是通过压缩后以.sra文件格式来保存的,SRA数据库可以用于搜索和展示SRA项目数据,包括SRA主页和 Entrez system,由 NCBI 负责维护。
ENA数据库:European Nucleotide Archive:隶属EBI (European Bioinformatics Institute),功能等同SRA,并且对保存的数据做了注释,界面相对于SRA更友好,对于有数据需求的研究人员来说,ENA数据库最诱人的点应该是可以直接下载fastq (.gz)文件,由 EBI 负责维护。
两者在主要功能方面非常类似,同时数据互通。
需要获取他人发表的公开测序数据,来帮助自己的研究领域,下载.sra文件是为了获取该sra相对应的fastq或者sam文件,通过文件格式转换就可以和自己的pipeline对接上,用于直接分析,所以:
第一步,我们需要到SRA或者ENA上搜索我们选择好的SRR号或者SRS号或者SRP号,先在ENA上搜索,如没有再去SRA上搜索,因为ENA下载比SRA快。
第二步,下载数据,从 SRA 数据库下载数据有多种方法。可以用ascp快速的来下载 sra 文件,也可以用wget或curl等传统命令从 FTP 服务器上下载 sra 文件(但是wget和curl下载的sra文件有时候会不完整),另外NCBI的sratoolkit 工具集中的prefetch、fastq-dump和sam-dump也支持直接下载,另外biostar handbook中有一个wonderdump脚本也方便下载数据,我以前还用过迅雷下载sra文件,直接得到sra的链接,迅雷下载。
高通量数据分析时,需要从公共数据库如 NCBI、EBI 下载他人提交的高通量测序数据。NCBI SRA 数据库下载的方式有很多种,普通用户可能常用 Entrez 网页版 检索并下载,但对于大批量高通量数据用命令行可以简化复杂的鼠标操作,实现后台自动化下载。这里介绍一些常用的工具和快速方法。
测序仪 basecalling 后转换成 fastq 格式的数据,包含序列信息和序列质量信息。而为了减少数据储存空间通常会进一步压缩成 .gz 格式。NCBI 开放高通量测序公共数据库后,为了进一步压缩文件大小,优化网络带宽,采用了 .sra 格式的文件。我们一般从 NCBI SRA 数据库下载的高通量测序数据均为 SRA 数据,需要通过 sra-tools 中的工具转换成 fastq 格式。SRA 是 Sequence Read Archive 的首字母缩写。
基本概念
SRA 与 Trace 最大的区别是将实验数据与 metadata(元数据)分离。metadata 是指与测序实验及其实验样品相关的数据,如实验目的、实验设计、测序平台、样本数据(物种,菌株,个体表型等)。metadata可以分为以下几类:
- Study:accession number 以 DRP,SRP,ERP 开头,表示的是一个特定目的的研究课题,可以包含多个研究机构和研究类型等。study 包含了项目的所有 metadata,并有一个 NCBI 和 EBI 共同承认的项目编号(universal project id),一个 study 可以包含多个实验(experiment)。
- Sample:accession number以 DRS,SRS,ERS 开头,表示的是样品信息。样本信息可以包括物种信息、菌株(品系) 信息、家系信息、表型数据、临床数据,组织类型等。可以通过Trace来查询。
- Experiment:accession number 以 DRX,SRX,ERX 开头。表示一个实验记载的实验设计(Design),实验平台(Platform)和结果处理(processing)三部分信息。实验是 SRA 数据库的最基本单元,一个实验信息可以同时包含多个结果集(run)。
- Run:accession number 以 DRR,SRR,ERR 开头。一个 Run 包括测序序列及质量数据。
- Submission:一个 study 的数据,可以分多次递交至 SRA 数据库。比如在一个项目启动前期,就可以把 study,experiment 的数据递交上去,随着项目的进展,逐批递交 run 数据。study 等同于项目,submission 等同于批次的概念。
常用工具
命令行下下载 SRA 常用工具:
- sra-tools:NCBI 官方开发的 SRA 工具,目前最新版为 2.10,注意:只能在 Linux 下运行,下载最大数据不超过20G。
- asperasoft:IBM 开发的快速下载工具。目前 NCBI 计划将 SRA 数据移植到云平台空间,可能将来不再允许 aspera 下载。目前 EBI ENA 还是可以使用。
- entrez-direct:NCBI官方开发的用于命令行的 entrez 工具。
sra-tools 是 NCBI 官方开发的 SRA 工具集,也是处理 SRA 数据处理的必备软件。常用的工具包括:prefetch,fastq-dump,vdb-config等。目前最新版的 sra-tools 为 v2.10,版本更新会带来功能上的变化,如果希望获得最新的功能建议将版本更新到 v2.10。
SRA Toolkit - prefetch 快速下载NCBI SRA数据
#1. sratoolkit 配置
#1.1 sratoolkit 下载
首先,下载最新发布的sratoolkit(基于自己的系统选择版本):
- CentOS Linux 64 bit architecture
- Ubuntu Linux 64 bit architecture
- MacOS 64 bit architecture
- MS Windows 64 bit architecture
$ wget https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.9.6/sratoolkit.2.9.6-ubuntu64.tar.gz
$ tar zxvf sratoolkit.2.9.6-ubuntu64.tar.gz
$ cd sratoolkit.2.9.6-ubuntu64
#加入环境路径
$ echo 'export export PATH=$PATH:Y/home/ljh2/biosoft/sratoolkit.2.9.6-ubuntu64/bin' >> ~/.bashrc
$ source ~/.bashrc
#1.2 检查sratoolkit 的配置
$ prefetch -V
prefetch : 2.9.6
#2 sratoolkit 使用
#2.1 prefetch 下载SRA数据
使用命令行下载SRA、dbGaP和ADSP数据
- 以
SRP193866数据为例
$ prefetch SRR8956151
2019-04-29T09:11:25 prefetch.2.9.6: 1) Downloading 'SRR8956151'...
2019-04-29T09:11:25 prefetch.2.9.6: Downloading via https...
2019-04-29T09:13:19 prefetch.2.9.6: https download succeed
2019-04-29T09:13:19 prefetch.2.9.6: 1) 'SRR8956151' was downloaded successfully
2019-04-29T09:13:19 prefetch.2.9.6: 'SRR8956151' has 0 unresolved dependencies
- 下载完成之后,SRR8956151.sra 保存在目录:
/ncbi/public/sra
$ sudo updatedb
$ locate SRR8956151.sra
~/ncbi/public/sra/SRR8956151.sra
#2.2 prefetch 批量下载数据
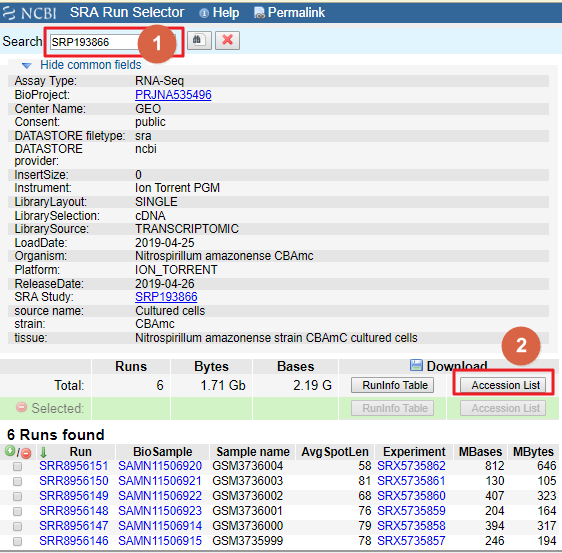
- SRA Run Selector 搜索
SRP193866信息, 点击Accession List获取Run Accessions: SRR_Acc_List.txt
SRA Run Selector
- SRR_Acc_List.txt
SRR8956146
SRR8956147
SRR8956148
SRR8956149
SRR8956150
SRR8956151
prefetch --option-file SRR_Acc_List.txt
prefetch -h查看帮助发现:
如果安装了aspera,prefetch默认调用aspera(Aspera 的核心是 fasp传输专利技术):
-t|--transport <value> Transport: one of: fasp; http; both. (fasp
only; http only; first try fasp (ascp), use
http if cannot download using fasp).
Default: both
#2.3 IBM Aspera 高速数据传输功能
- 以最快的速度发送和共享大型文件和数据集。
- 快速传输、分发和同步大型文件和数据集。
- 全面自动统筹、监控和控制数据传输和工作流程。
- 通过互联网近乎零延迟地交付任何规模的数据,以及几乎无限比特率的视频。
#2.4 Aspera 安装
$ wget https://download.asperasoft.com/download/sw/connect/3.9.1/ibm-aspera-connect-3.9.1.171801-linux-g2.12-64.tar.gz
$ tar zxvf ibm-aspera-connect-3.9.1.171801-linux-g2.12-64.tar.gz
$ bash ibm-aspera-connect-3.9.1.171801-linux-g2.12-64.sh
Installing IBM Aspera Connect
Install complete.
#加入环境路径
$ echo 'export PATH=$PATH:/home/lujunhui2/.aspera/connect/bin' >> ~/.bashrc
$ source ~/.bashrc
#2.5 prefetch 调用Aspera
- 数据下载速度真的快
$ prefetch SRR8956151
2019-04-29T16:20:27 prefetch.2.9.6: 1) Downloading 'SRR8956151'...
2019-04-29T16:20:27 prefetch.2.9.6: Downloading via fasp...
SRA 数据库下载
NCBI 数据库目前有3个地址
- ncbi.public
- aws s3
- google cloud public
注:用prefetch无法下载超过20G的数据,我们可以用wget
缺点:wget下载数据太慢了,推荐用aria2c下载网络数据能够极大的提高下载SRR数据的速度。
aria2
是一个自由、开源、轻量级多协议和多源的命令行下载工具。它支持 HTTP/HTTPS、FTP、SFTP、 BitTorrent 和 Metalink 协议。aria2 可以通过内建的 JSON-RPC 和 XML-RPC 接口来操纵。aria2 下载文件的时候,自动验证数据块。它可以通过多个来源或者多个协议下载一个文件,并且会尝试利用你的最大下载带宽。默认情况下,所有的 Linux 发行版都包括 aria2,所以我们可以从官方库中很容易的安装。一些 GUI 下载管理器例如 uget 使用 aria2 作为插件来提高下载速度。
Aria2 特性
- 支持 HTTP/HTTPS GET
- 支持 HTTP 代理
- 支持 HTTP BASIC 认证
- 支持 HTTP 代理认证
- 支持 FTP (主动、被动模式)
- 通过 HTTP 代理的 FTP(GET 命令行或者隧道)
- 分段下载
- 支持 Cookie
- 可以作为守护进程运行。
- 支持使用 fast 扩展的 BitTorrent 协议
- 支持在多文件 torrent 中选择文件
- 支持 Metalink 3.0 版本(HTTP/FTP/BitTorrent)
- 限制下载、上传速度
1) Linux 下安装 aria2
我们可以很容易的在所有的 Linux 发行版上安装 aria2 命令行下载器,例如 Debian、 Ubuntu、 Mint、 RHEL、 CentOS、 Fedora、 suse、 openSUSE、 Arch Linux、 Manjaro、 Mageia 等等……只需要输入下面的命令安装即可。对于 CentOS、 RHEL 系统,我们需要开启 uget 或者 RPMForge 库的支持。
# yum install aria2[Fedora 22 和 之后的系统]
# dnf install aria2[对于 suse 和 openSUSE]
# zypper install wget[Mageia]
# urpmi aria2[对于 Arch Linux]$ sudo pacman -S aria2
2) 下载单个文件
下面的命令将会从指定的 URL 中下载一个文件,并且保存在当前目录,在下载文件的过程中,我们可以看到文件的(日期、时间、下载速度和下载进度)。
# aria2c https://download.owncloud.org/community/owncloud-9.0.0.tar.bz2[#986c80
3)续传未完成的下载
当你遇到一些网络连接问题或者系统问题的时候,并将要下载一个大文件(例如: ISO 镜像文件),我建议你使用 -c 选项,它可以帮助我们从该状态续传未完成的下载,并且像往常一样完成。不然的话,当你再次下载,它将会初始化新的下载,并保存成一个不同的文件名(自动的在文件名后面添加 .1 )。注意:如果出现了任何中断,aria2 使用 .aria2 后缀保存(未完成的)文件。
# aria2c -c https://download.owncloud.org/community/owncloud-9.0.0.tar.bz2[#db0b08
4) 每个主机使用两个连接来下载
默认情况,每次下载连接到一台服务器的最大数目,对于一条主机只能建立一条。我们可以通过 aria2 命令行添加 -x2(2 表示两个连接)来创建到每台主机的多个连接,以加快下载速度。
# aria2c -x2 https://download.owncloud.org/community/owncloud-9.0.0.tar.bz2[#ddd4cd
fastq-dump: 一个神奇的软件
现在可以用
fasterq-dump, 速度更快,请阅读2202年了,还用fastq-dump,快换fasterq-dump吧
做生信的基本上都跟NCBI-SRA打过交道,尤其是fastq-dump大家肯定不陌生.NCBI的fastq-dump软件一直被大家归为目前网上文档做的最差的软件之一",而我用默认参数到现在基本也没有出现过什么问题,感觉好像也没有啥问题, 直到今天看到如下内容, 并且用谷歌搜索的时候,才觉得大家对fastq-dump的评价非常很到位.
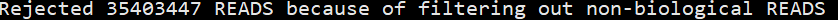
过滤
我们一般使用fastq-dump的方式为
fastq-dump /path/to/xxx.sra
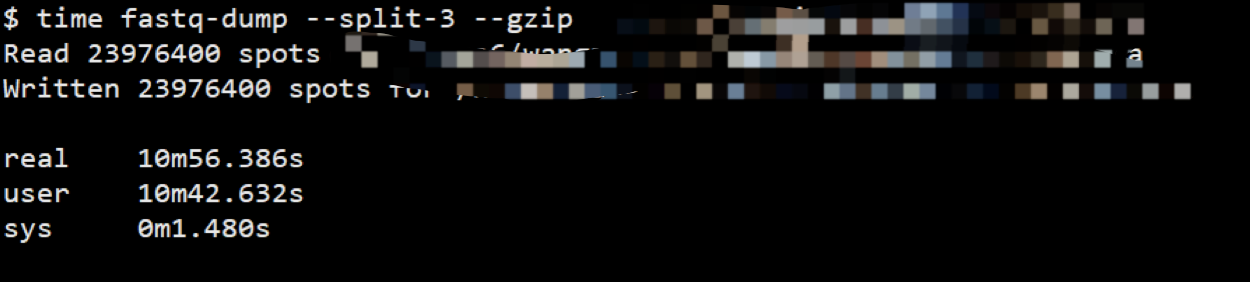
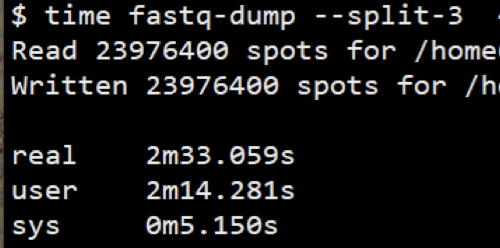
但是这个默认使用方法得到结果往往很糟, 比如说他默认会把双端测序结果保存到一个文件里, 但是如果你加上--split-3之后, 他会把原来双端拆分成两个文件,但是原来单端并不会保存成两个文件. 还有你用--gzip就能输出gz格式, 能够节省空间的同时也不会给后续比对软件造成压力, 比对软件都支持,就是时间要多一点。
–gzip时间
没有gzip时间
但是很不幸运,这些东西在官方文档并没有特别说明,你只有通过不断的踩坑才能学到这些小知识。
我建议尽量去EMBL-EBI去下载原始数据,而不是这种神奇的sra格式,尽管有一些下载的数据其实就是从SRA解压而来。
不过要用fastq-dump,那就介绍几个比较重要的参数吧。我会按照不懂也加,不懂别加,有点意思,没啥意义这三个级别来阐述不同参数的重要级.
太长不看版
如果你不想了解一些细节性内容,用下面的参数就行了
fastq-dump --gzip --split-3 --defline-qual '+' --defline-seq '@\$ac-\$si/\$ri' SRR_ID
# 建议加别名
alias fd='fastq-dump --split-3 --defline-qual '+' --defline-seq '@\\\$ac-\\\$si/\\\$ri' '
数据格式
不懂也加: reads拆分
默认情况下fastq-dump不对reads进行拆分, 对于很早之前的单端测序没有出现问题.但是对于双端测序而言,就会把原本的两条reads合并成一个,后续分析必然会出错.
常见的参数有三类:
--split-spot: 将双端测序分为两份,但是都放在同一个文件中--split-files: 将双端测序分为两份,放在不同的文件,但是对于一方有而一方没有的reads直接丢弃--split-3: 将双端测序分为两份,放在不同的文件,但是对于一方有而一方没有的reads会单独放在一个文件夹里
关于遇到的Rejected 35403447 READS because of filtering out non-biological READS就是因为原来是SE数据,但是用--split-3当作PE数据处理,出现的问题. 看起来好像有问题,但是对后续结果分析没有太多影响.
因此,对于一个你不知道到底是单端还是双端的SRA文件,一律用--split-3.
没事别加: read ID
默认双端测序数据拆分后得到两个文件中同一个reads的名字是一样的,但是加上-I | --readids之后同一个reads的ID就会加上.1和.2进行区分.举个例子
| 是否有-I参数 | ID 1 | ID 2 |
|---|---|---|
| 无 | @SRR5829230.1 1 length=36 | @SRR5829230.1 1 length=36 |
| 有 | @SRR5829230.1.1 1 length=36 | @SRR5829230.1.2 1 length=36 |
问题来了, 明明已经可以通过ID后面的"1"和"2"来区分ID, 加这个参数干嘛. 加完之后还会让后续的BWA报错.所以,没事千万别加
有点意义: 原始格式
默认情况下输出的文件的ID都是SRR开头,但其实原始数据名字不是这样子,比如说@ST-E00600:143:H3LJWALXX:1:1101:5746:1016 2:N:0:CCTCCTGA,@HWI-ST620:248:HB11HADXX:2:1101:1241:2082#0/1这种. 如果你想看到那种格式,而不是SRR,你需要怎么做呢?
可以通过如下三个选项进行修改
F|--origfmt: 仅保留数据名字--defline-seq: 定义readsID的显示方式--defline-qual: 定义质量的显示方式
其中fmt按照如下要求定义
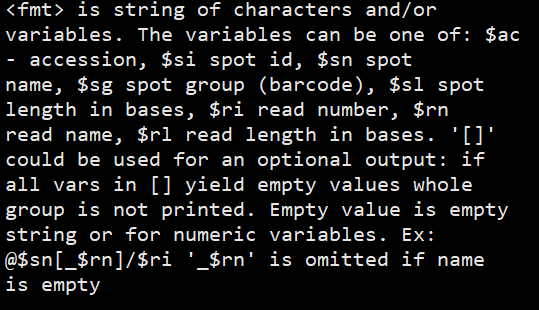
fmt的写法
虽然看起来有点意思,但是对最后的分析其实没啥帮助.
没啥意义: fasta输出
如果下游分析只需要用到fasta文件,那么用--fasta就行. 当然了也有很多方法能够把fastq转换成fasta,比如说samtools.
过滤
我觉得这部分的参数都没有意义, 毕竟完全可以用专门的质控软件处理reads,不过--skip-technical,是唯一比较重要.
- 根据ID:
-N-X - 根据长度:
-M - 多标签序列:
--skip-technical, 这个是唯一有点意思的,就是说如果你原来建库测序使用了多个标签来区分序列, 默认不会输出这个标签. 但是如果不输出标签,我们怎么区分呢? 所以一定要显示声明
有点意思: 输出方式
这部分参数也很重要, 选择是否压缩,还是直接输出到标准输出
--gzip,--bzip2: 压缩方式-Z | --stdout: 输出到标准输出-O|--outdir: 输出到指定文件夹

旨在为数千万中国开发者提供一个无缝且高效的云端环境,以支持学习、使用和贡献开源项目。
更多推荐



 25
25 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)