




- @yijiaobani
简介
该用户还未填写简介
擅长的技术栈
可提供的服务
暂无可提供的服务
大家好,我是邓飞。Positron出来好久了,但是我一直用Rstudio了,之前在Rstudio中试用过AI补全代码,太糟心了。就去掉了AI模块。AI变成我用的是Trae,但是一直让我排队,免费的,也可以理解。我的DeepSeek API一直用claude,倒也平静。最近,我想跑一些python代码,想用Rstudio跑一下,结果各种麻烦,听说Positron跑python更流畅,就下载试一下,果
1. 为何要计算BLUE值?一年多点或者多年多点的植物数据中,一个基因型(品种)往往有多个表型数据,但只有一个基因型,在GWAS关联分析中,就需要一个基因型对应一个表型数据。之所以有多个表型数据,或者是多个重复,或者是多个地点的数据,或者是多个年份的数据,如何计算得到一个表型数据呢?可以使用多个表型值的平均值,作为品种的表型值,现在有更好的方法:BLUE值。2. 为何使用BLUE值?一般...
1. 背景之前写过一篇博客, 介绍领导安利我哔哩哔哩的故事, 介绍到我将我从YouTube上收集的关于混合线性模型, 关于GWAS, 关于GS, 关于农业数据分析相关的视频, 上传到了哔哩哔哩上面. 今天我们看一下介绍多年多点遗传力及BLUP值计算的视频内容. 阅读原文可以查看视频, 这里我用文字和代码进行重演.2. 本次微信文的目标获得一个多年多点的数据计算品种性状的遗传力计算每个品...
3,数据集的组织和存储方式应便于重新分析。5,使用考虑空间趋势的混合模型增加了显着QTL的数量,这可能是由于性状的狭义遗传力和GWAS的统计能力增加的结果。上面是数据是否清洗,检测的结果,可以看到,第10条染色体在数据清洗后,没有了显著性位点,这些位点可能是假阳性。3,另外,建议可以识别异常值,但是不能删除,异常值也是有价值的信息,可以分别测试不同的结果。2,表型数据,使用原始数据,blup值,平
1, 编者自语H矩阵作为一步法的入门技术, 是需要掌握的, 本文以一篇文献为例, 介绍如何从头构建H矩阵. 文章包括H矩阵推导过程和代码实现.2, H矩阵定义基因组选择中, GBLUP的一个挑战是, 在参考群构建时, 需要两步, 第一步根据系谱和表型数据, 计算出伪数据(pseudo-data)(比如, 根据系谱计算公牛的女儿产奶偏差作为表型值, 因为公牛没有产奶数据), 然后用基因组信息进...
这篇博客,用之前GWAS教程中的示例数据,把数据分为Base数据和Target数据,通过plink运行二分类的logistic模型进行GWAS分析,然后通过PRSice-2软件,进行PRS分析。最终,选出最优SNP组合,并计算Target的PRS得分,主要结果如下:最适合的SNP个数是133个,R2位0.232258,P值为0.014上面数据中,个体的PRS为正值,说明风险高,为负值,说明风险低。
因此, 比较好的方式是,在one-stage中,将地点,年份,区组作为随机因子,将品种作为固定因子,计算BLUE值。对于纵向数据(比如不同胎次的产仔数,不同时期的剪毛量),对于一般的GLM模型,MLM模型,需要用平均值或者BLUE值作为表型值。对于植物数据,特别是随机区组的数据,一年多点的数据,一个基因型ID对应多个表型值,这些表型值如何利用呢?另外,就是进行汇总统计,看一下最大值,最小值,平均值
令人击节的一段话:随着全基因组选择统计模型的不断改进优化,模型的稳定性及准确性不断提高,但是依然面临两个重要的挑战,即计算准确性和计算效率;直接法(GBLUP为代表)计算效率较高,但是计算准确性略差于间接法(BayesB为代表),虽然学者对直接法进行了改进,但是由于改进的策略中人为设定参数较多,因此模型的预测准确性受主观因素影响较大;间接法计算准确性较高,但是由于参数求解过程中计算量庞大,且无法.
这里,模拟一个plink文件的数据,8个样本,8个SNP位点,通过手动Excel计算样本杂合度和位点杂合度,比较plink计算杂合度的方法。1. 模拟数据ped数据:$ cat a.pedFAMILY1ID1000-9CCCCAAGGAGGGGGGCFAMILY1ID2000-9
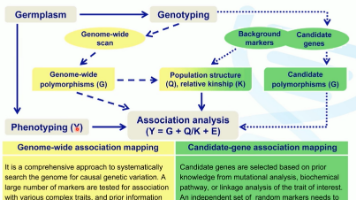
1, 序言这篇是基因组选择的理论加实践,因为我看到一句话,Talk is cheap. Show me the code,很有感触,有感而写。使用的包是R的sommer和asreml,其实强健的还是成熟的软件,比如DMU,BLUPF90,PIBLUP,ASreml等,但sommer作为基本功能的演示,非常合适。2, 定义基因组选择(Genomic Selection, GS), 利用覆盖全基...