




- @Igenebook
简介
该用户还未填写简介
擅长的技术栈
可提供的服务
暂无可提供的服务
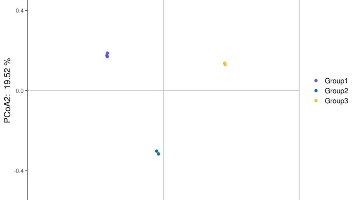
爱基百客云平台推出PCoA分析工具,帮助用户快速实现Beta多样性可视化。该工具支持多种距离算法(如Bray-Curtis、Jaccard等),适用于处理非正态分布的多维数据。用户只需上传基因表达矩阵和分组文件,选择距离计算方法,即可自动生成三种可视化结果(点图、置信区间图和样本名图)。平台提供38款零代码分析工具,操作简单,支持PDF/PNG格式输出,满足微生物组学、转录组等研究的降维分析需求。
近期,重庆大学雷明星教授团队联合美国南加州大学钟正明院士团队、陆军军医大学烧伤研究所罗高兴教授团队在国际知名期刊Nature Communications发表研究论文。该研究以皮肤类器官为研究模型,整合了scRNA-seq、ChIP-seq、ATAC-seq、RNA-seq等多组学和分子功能实验,揭示了低氧如何驱动皮肤类器官从球状聚合体向平面化皮肤结构转变。

植物单细胞测序研究者在获得细胞分群和注释后,常面临解析细胞间通讯网络的挑战。厦门大学郑海雷课题组开发的PlantPhoneDB数据库填补了植物领域配体-受体互作资源的空白,支持拟南芥、水稻等5个物种的单细胞通讯分析。该开源数据库提供配体-受体配对信息、R分析代码和可视化工具,可生成通讯热图、弦图等结果,量化细胞间信号流向与强度。通过整合表达数据与L-R词典,研究者能系统解析"谁在发送信号

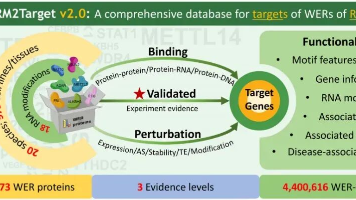
中山大学团队开发的RM2Targetv2.0数据库为RNA修饰研究提供了重要工具,系统收录了18种RNA修饰相关的Writers、Erasers和Readers蛋白的靶基因信息。该数据库整合了440多万条WER-靶标关联数据,涵盖20个物种的973种细胞系/组织,包含已验证、结合和扰动三类实验证据。最新版本创新性地采用大语言模型辅助数据审阅,并提供多维注释信息,包括修饰图谱、分子互作网络和临床疾病
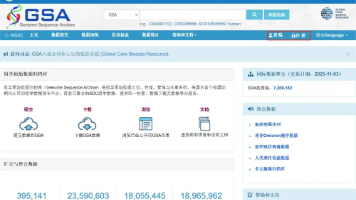
相比以往大家熟知的NCBI,GSA数据库符合国家数据安全标准,而且可以选择中文界面,对国人来说使用感受更加友好。如果是人类遗传资源相关组学原始数据,遵从《中华人民共和国人类遗传资源管理条例》总则规定,建议您将数据提交到GSA-Human数据库,以实现人类遗传资源数据的受控访问、保障人类遗传源数据的安全性。GSA for Human数据库网址:https://ngdc.cncb.ac.cn/gsa-
泛基因组的提出弥补了这一不足,它通过整合多个个体的基因组信息,构建出包含核心基因组(所有个体共有的基因)和可变基因组(部分个体特有的基因)的综合基因组框架,从而揭示种群内的遗传多样性和进化规律。基于泛基因组的优势,可以想见未来泛基因组会成为参考基因组的新标准。基因组包含生物体的全套遗传信息,研究中通常会将一个物种中重要的品系或者最先测出的基因组作为参考基因组,并以此为基础进行个体或群体水平的遗传变

一文教您如何进行DAP-seq的数据挖掘,筛选验证位点:从分析前思路→分析思路→如何筛选验证位点。
总之,PlantPAN为我们提供了一个强大的植物启动子和转录因子动态调控的信息,我们可以查找不同植物的基因ID以及对应的注释信息以及启动子和调控信息,还可以输入基因集,去探索基因集的调控关系,还可以进行跨物种分析,得到不同物种的基因启动子区域相似性信息以及转录因子结合位点信息、CpG岛和串联重复信息。点击Network construction的view可以看出来,这一组基因分别被AT2G4659

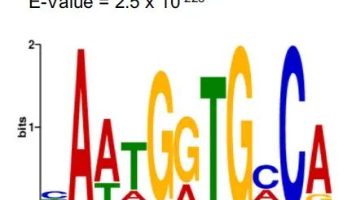
在NGS背景下,Motif(基序)是指DNA/RNA序列中反复出现的、具有生物学功能的短保守序列模式(通常5-20bp),代表转录因子(TF)等蛋白质的结合位点。在NGS(尤其是ChIP-seq)中,Motif分析是连接“蛋白结合位点”与“具体蛋白”的关键步骤,常与peak calling后的序列提取与注释配套进行。将实验序列与数据库中的已知motif(如JASPAR)进行比对,评估哪些转录因子可
深度学习作为一种极具潜力的多功能工具,在图像处理及大数据分析领域展现出显著优势。
