- @Asa12138
简介
该用户还未填写简介
擅长的技术栈
可提供的服务
暂无可提供的服务
与此同时,AlphaFold革命及超过2亿个蛋白质结构模型的发布,也推动了基于结构的聚类工具(如FoldSeek)的发展,进一步丰富了蛋白家族的多样性。其SP分数在所有工具中下降幅度最小,显示出最佳的稳定性。值得注意的是,FAMSA2的Medoid树模式所需时间比次优的Clustal Omega少227倍,而唯一具有可比执行时间的Kalign所产生的比对质量明显较低。为了测试比对工具在包含无关序列

Mermaid是一个基于 JavaScript 的图表绘制工具,可渲染 Markdown启发的文本定义以动态创建和修改图表,允许使用文本和代码创建图表和可视化。
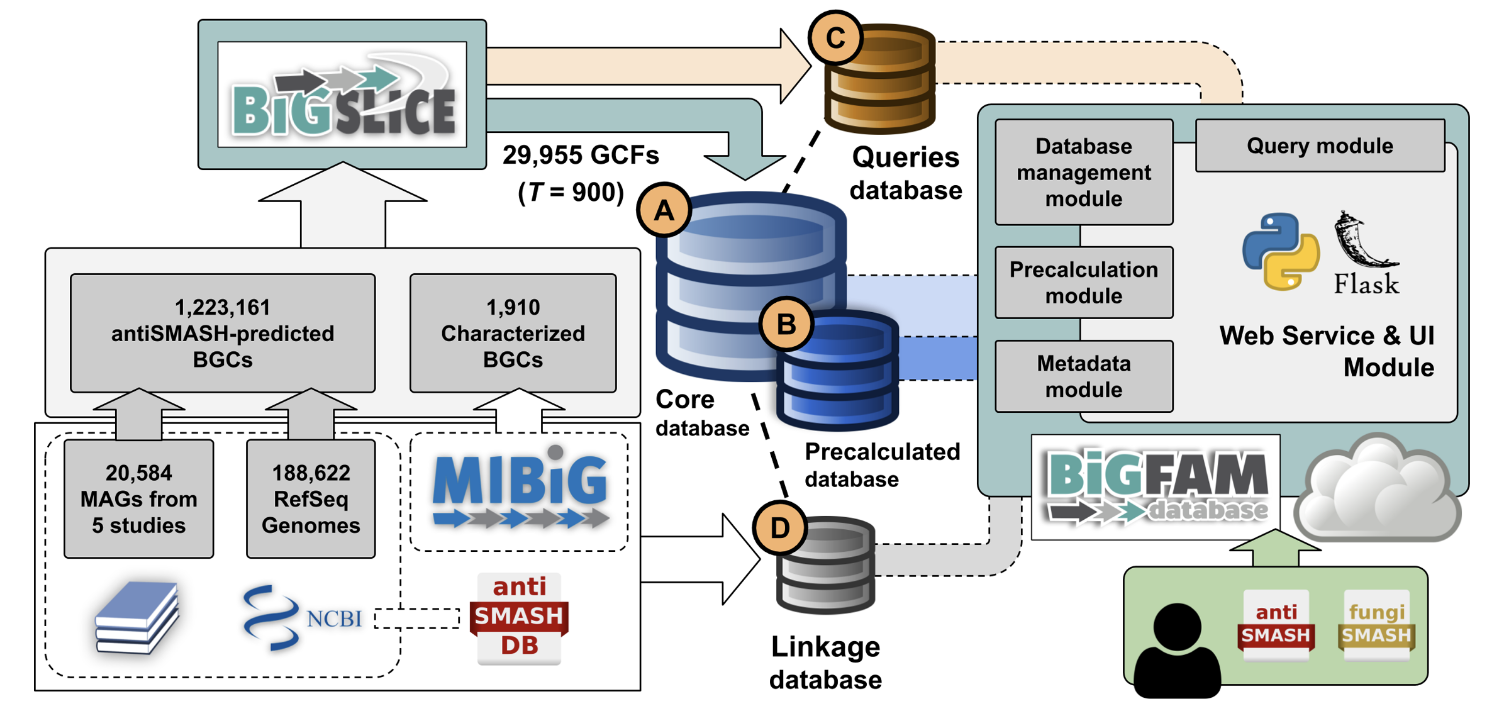
通过将从当前可用的基因组和 MAG 中识别的大规模全球 BGC 集合作为数据源,BiG-FAM 提供了可探索的微生物次生代谢多样性“图集”,以浏览和搜索跨类群的生物合成多样性。
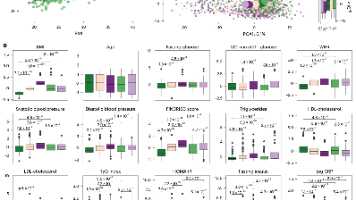
为了在解决共线性的同时提高模型简洁性,研究人员使用与BMI最严格相关的267种代谢物训练了一个岭回归模型,所得的预测值即为metBMI。在调整了年龄、性别和BMI后,提取每个参与者的metBMI残差。残差大于+2.5的个体被定义为高metBMI(HmetBMI),小于-2.5的个体被定义为低metBMI(LmetBMI),各占队列约10%。
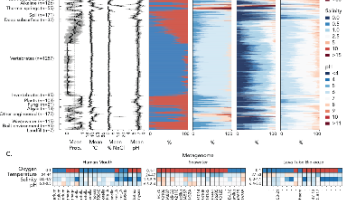
结果显示,海洋MAGs的防御库有限,这可能源于多种因素:开放海洋中普遍存在基因组精简的类群、浮游生活方式的普遍性、低细胞密度、水平基因转移频率较低,以及现有防御系统HMM模型主要基于可培养细菌(与全球海洋微生物组亲缘关系较远)开发。在防御岛中,多个防御家族的过表达并未转化为与防御组其余部分更高的共定位可能性,这提示了防御岛内部分选定基因家族间可能存在未被认识的上位相互作用或功能多样化。在不同环境中
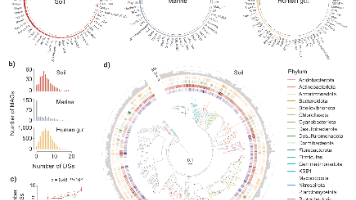
针对其中最具潜力的核糖体合成和翻译后修饰肽类生物合成基因簇,研究人员开发了基于蛋白质大语言模型的深度学习框架,用于从EEMC中预测基因组编码的、非毒性的候选抗菌肽,共鉴定出3,032个候选肽。综上所述,EEMC为发掘新的微生物谱系和生物合成能力奠定了资源基础,其整合人工智能与实验验证的管线,为未来的药物发现和生物技术应用提供了强大的工具与洞见。其次,大多数BGCs,尤其是来自宏基因组组装基因组的B
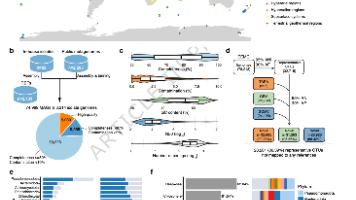
目前微生物学和环境微生物学中,有很大一部分微生物尚未被成功培养(即“不可培养”微生物)。这部分原因在于难以确定其适宜的培养条件(如氧气要求、最适温度、pH、盐度等)。虽然有方法可以通过基因组中注释出的代谢基因来预测碳源或能量代谢方式,但对于诸如氧气耐性、温度、pH、盐度等“培养条件参数”,基于功能基因的预测比较困难,特别是在基因注释不完整或未知功能基因广泛存在的情况下。如果能从基因组层面(不依赖于
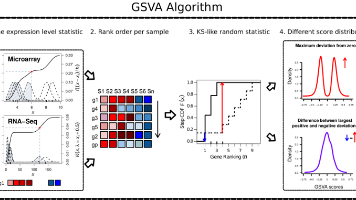
基因集变异分析 (GSVA) 通过将输入的基因×样本表达数据矩阵转换为相应的基因集×样本表达数据矩阵,从而提供通路活性的估计。生成的结果表达数据矩阵随后可用于经典的差异表达、分类、生存分析、聚类或相关性分析等方法,进行通路中心的分析。人们还可以在通路与其他分子数据类型(如 microRNA 表达或结合数据、拷贝数变异 (CNV) 数据或单核苷酸多态性 (SNPs))之间进行样本间的比较。GSVA
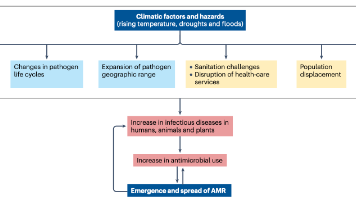
研究表明,58%的人类致病性疾病对气候变化敏感,通过千余种传播途径(媒介传播、水源性、气源性等)加剧疾病负担。气温升高不仅增加感染风险,还直接关联耐药性发展。低收入国家需平衡抗生素管控与可及性,减少50%全球用量仅能降低AMR 2.1%,而结合卫生系统改善的综合策略效果提升至5.1%。研究表明,细菌为适应高温进化出的RNA聚合酶突变,可同时导致对利福平的交叉耐药,提示气候适应可能驱动AMR进化。热
更重要的是,Evo 2能够生成整个细胞器(如人类线粒体)的完整序列,生成的序列包含了正确数量的编码序列、tRNA和rRNA基因,并保持了正确的基因排列顺序和密码子使用偏好。Evo 2强调通用能力,而非针对特定任务的优化,它在生物序列建模领域树立了一个重要里程碑,为与中心法则所有“模态”(DNA、RNA、蛋白)相关、从分子到基因组尺度、并可跨越所有生命领域泛化的预测与设计任务,奠定了广泛的基础。值得