iMetaOmics | 中国药科大学徐文波开发叶绿体基因组数据分析软件
点击蓝字 关注我们CPStools:专为叶绿体基因组设计的分析软件研究论文●原文链接DOI: https://doi.org/10.1002/imo2.25●2024年8月23日,中国药科大学徐文波团队在iMetaOmics在线发表了题为“CPStools: a package for analyzing chloroplast genome sequences”的文章。●本研究开发的CPSt..
点击蓝字 关注我们
CPStools:专为叶绿体基因组设计的分析软件

研究论文
● 原文链接DOI: https://doi.org/10.1002/imo2.25
●2024年8月23日,中国药科大学徐文波团队在iMetaOmics在线发表了题为“CPStools: a package for analyzing chloroplast genome sequences”的文章。
● 本研究开发的CPStools是一款专用于叶绿体基因组分析的用户友好型软件。CPStools集成了10个子命令,通过简化输入要求并自动化处理复杂流程,显著提高了叶绿体基因组分析的效率和准确性。
● 第一作者:黄丽瑾、于换喜
● 通讯作者:徐文波(xwb7533@163.com)
● 主要单位:中国药科大学中药学院、生态环境部南京环境科学研究所
亮 点
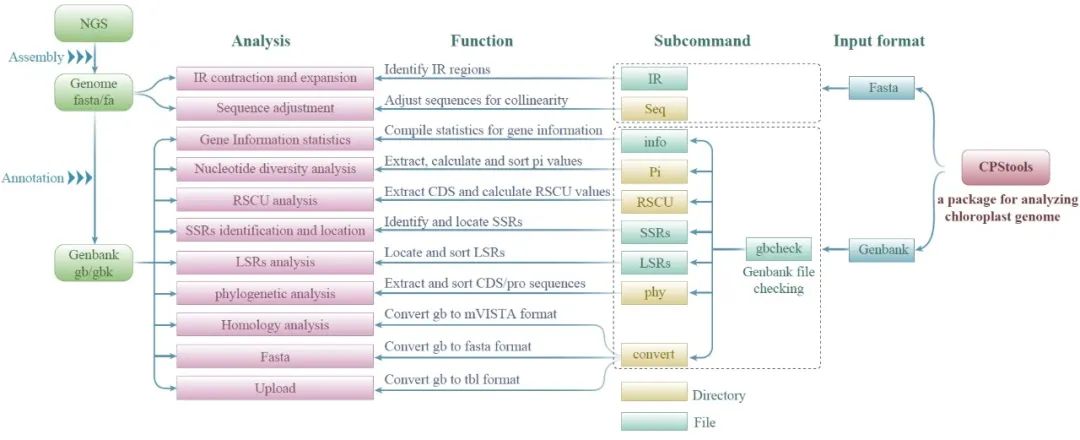
● CPStools集成了叶绿体基因组中的10个关键分析;
● CPStools使复杂的分析一键化,提高了叶绿体基因组分析效率和准确性;
● CPStools提供了友好的用户界面,使其成为相关研究人员的重要工具。
摘 要
CPStools是一款专用于叶绿体基因组分析的用户友好型软件。它集成了10个核心功能,包括注释文件检查、基因信息统计、四分体结构鉴定、序列调整、核苷酸多样性分析、密码子偏好性分析、简单重复序列分析、长重复序列统计、系统发育分析和格式转换。CPStools接受Fasta和Genbank格式的文件作为输入,自动化执行各分析流程,在提高准确性的同时也为研究人员节省了大量时间。CPStools还提供了界面友好、操作简便的使用体验,并附有详细的说明文档。
视频解读
Bilibili:https://www.bilibili.com/video/BV1zT4sebEAJ/
Youtube:https://youtu.be/UOvrcfk7xhg
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
叶绿体是绿色植物和藻类等真核自养生物进行光合作用的细胞器,其基因组通常为典型的环状四分体结构,包含小单拷贝区(Small Single Copy, SSC)、大单拷贝区(Large Single Copy, LSC)和两个反向重复区(Inverted Repeat, IR)。叶绿体基因组在系统分类和物种鉴定中具有重要的作用。随着高通量测序技术的发展,叶绿体基因组的测序与分析已成为常规操作。然而,目前的叶绿体基因组分析工具仍存在显著局限性。例如,MIcroSAtellite Identification(MISA)虽被广泛用于检测简单重复序列(Simple Sequence Repeats, SSRs),但其复杂的输出结果对缺乏经验的科研人员来说较难处理。CodonW用于计算相对同义密码子使用频率(Relative Synonymous Codon Usage, RSCU),但其输入文件的准备过程繁琐,需从多个Genbank文件中提取共有的蛋白编码序列(Protein-coding Sequences, CDS),并过滤掉短序列、假基因和重复序列,耗时较长。此外,使用Geseq和Geneious在识别叶绿体基因组四分体区域时,若IR区域存在跨区域的短片段,往往会生成不准确的鉴定结果。同时,目前缺乏专用于叶绿体基因组核酸多态性(Nucleotide Diversity, Pi)分析和共有CDS序列及蛋白序列提取的软件。
为了解决这些问题,我们开发了CPStools,该工具集成了10个子命令,每个命令都提供特定功能,克服了现有工具的局限性。通过简化输入要求并自动化处理复杂流程,CPStools显著提高了叶绿体基因组分析的效率和准确性。这种简化的方法不仅为研究人员节省了大量时间,还减少了错误发生的可能性,使CPStools成为叶绿体基因组研究中不可或缺的重要工具。
结 果
CPStools集成了叶绿体基因组分析中10个重要的功能。这些功能包括注释文件检查、基因信息统计、四分体结构鉴定、序列调整、核苷酸多样性分析、密码子偏好性分析、简单重复序列分析、长重复序列统计、系统发育分析和格式转换。利用CPStools对从NCBI下载的9个绞股蓝属植物叶绿体基因组序列进行了比较分析。使用注释文件检查功能,共鉴定出13个不以“ATG”为起始密码子的基因,主要为ndhD和psbL,这些基因在叶绿体基因组中通常为RNA编辑的热点区域。9个叶绿体基因组均注释出113个非冗余基因,唯独喙果绞股蓝(Gynostemma yixingense)在trnfM-CAU和trnM-CAU基因的注释中存在错误,这是研究人员常犯的一个注释错误。使用“IR”子命令对9条序列进行四分体结构鉴定,发现有2条不以LSC区域的第一个碱基作为序列起点。而在Geseq和Geneious中,鉴定结果表明这9条序列均以LSC区域的第一个碱基作为起点,当重复区短片段跨区域时,这两个软件往往无法进行准确的四分体区域鉴定(表S1)。结合共线性结果和“IR”鉴定结果,“Seq”子命令中的三种模式可轻松调整这些序列。使用Pi子命令进行核酸多态性分析,提取了110个共有基因和150个共有基因间区。经过多序列比对并计算pi值后,选择pi值较高的区域作为条形码区域,用于后续的物种鉴定和分子标记筛选(图1A)。GenBank格式转换为mVISTA格式后,也可以通过mVISTA进行准确的可视化分析(图1B)。在密码子偏好性分析中,CPStools可一键提取和过滤CDS序列,并计算RSCU值。经过多重过滤,绞股蓝保留了51个基因,其余8个物种均保留了52个基因(图1C)。在简单重复序列分析中,9个绞股蓝属物种中分别鉴定出44、55、52、58、37、62、47、54和45个SSRs,同时记录了这些SSRs在基因间隔区、内含子区和外显子区域中的位置(图1D,表S2)。所有分析均可在半小时内高精度完成,这极大地节省了研究人员的时间。
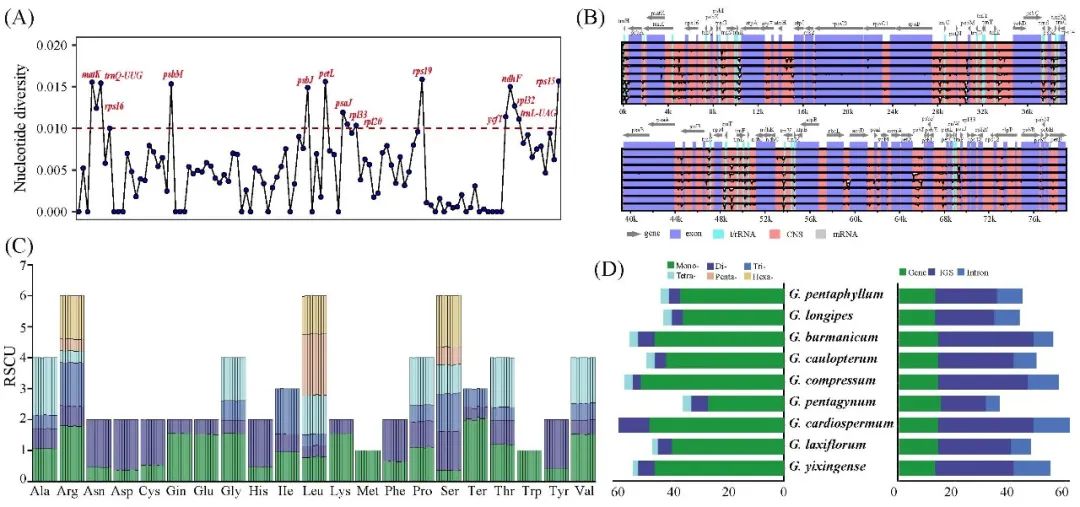
图1. CPStools分析结果与可视化
(A)共有基因核苷酸多样性分析;(B)mVISTA转换及部分可视化;(C)密码子偏好性分析;(D)简单重复序列鉴定及定位。
方 法
“gbcheck”功能提供了两种模式:自检模式和比对模式。在自检模式下,脚本会检查CDS基因,判断起始和终止密码子是否正确,并检查是否存在多个终止密码子。比对模式则通过识别基因注释中的差异基因数目来比较注释文件。“info”子命令提供基因数目、类型和外显子数量的统计分析。这两个功能可提高叶绿体基因组注释的准确性并生成详细的基因信息统计表。
大多数植物的叶绿体基因组测序数据在组装和重复区复制后,可形成典型的四分体结构。当重新解环为线性结构时,叶绿体基因组的起始位点可以选择任意位置,并且小单拷贝区有时可能出现反向重复现象。通过使用CPStools中的“IR”子命令,可以鉴定四分体结构。根据鉴定结果和共线性分析结果,可以选择“Seq”子命令中的三种模式进行序列调整,使叶绿体基因组以大单拷贝区的第一个碱基作为序列的起始位点,并确保小单拷贝区的方向与参考序列一致。
“Pi”分析用于检测序列的核酸多态性,其中高突变率的区域可作为物种鉴定的分子标记。然而,从注释文件中提取共有基因和基因间隔区序列的过程非常复杂,而使用DNAsp6计算pi值也较为耗时,因为它只能处理单个比对文件。考虑到整个叶绿体基因组中提取的共有序列通常超过200个,这一过程显得更加繁琐且费时。CPStools中“Pi”子命令可接受包含多个Genbank文件的文件夹作为输入。软件会解析每个文件,提取其中共有的基因区域和基因间区。通过多序列比对和pi值计算后,软件将按各区域在叶绿体基因组中的顺序进行排序,并绘制相应的图片,为下一步的分子标记筛选提供参考。

图2. CPStools中核酸多态性分析流程
密码子偏好性分析在研究叶绿体基因组中的密码子使用模式时具有重要意义,其分析流程主要包括过滤短序列、去除重复序列和假基因,并计算相对密码子的使用频率。CPStools接受包含多个Genbank文件的文件夹作为输入。首先,它解析Genbank文件并提取CDS序列。接着,根据设定的过滤阈值,对序列进行过滤,同时去除假基因和重复序列。最后,软件将过滤后的序列合并并计算RSCU值,并根据输入文件的数量绘制相应的统计图。
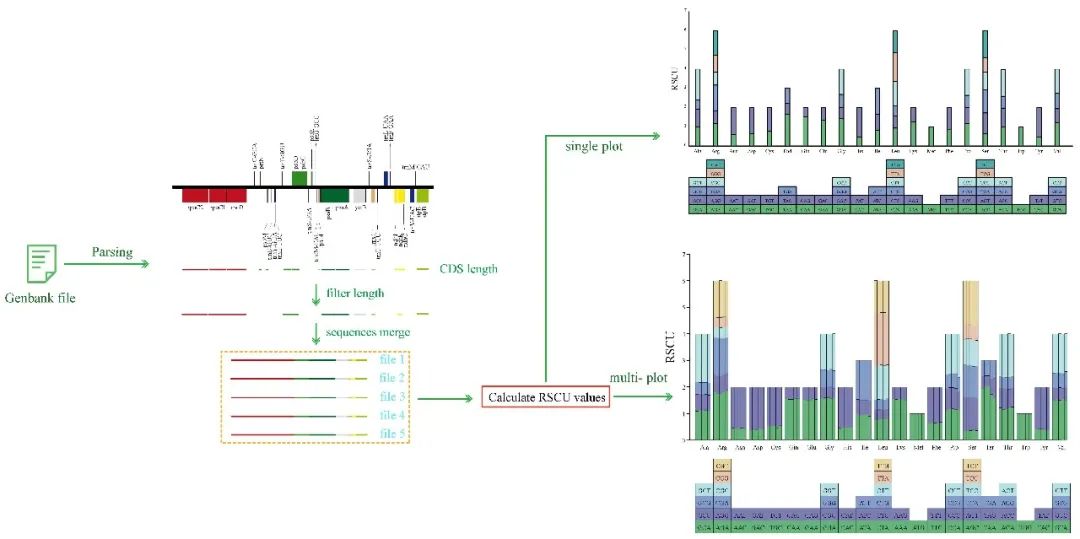
图3. CPStools中密码子偏好性分析流程
在简单重复序列分析中,CPStools接受Genbank文件作为输入。首先,它将Genbank文件转换为Fasta格式,并使用正则表达式鉴定序列中的简单重复序列。接着,CPStools解析Genbank格式文件,记录基因、内含子和基因间区的位置,并将鉴定出的简单重复序列映射到这三个区域中。
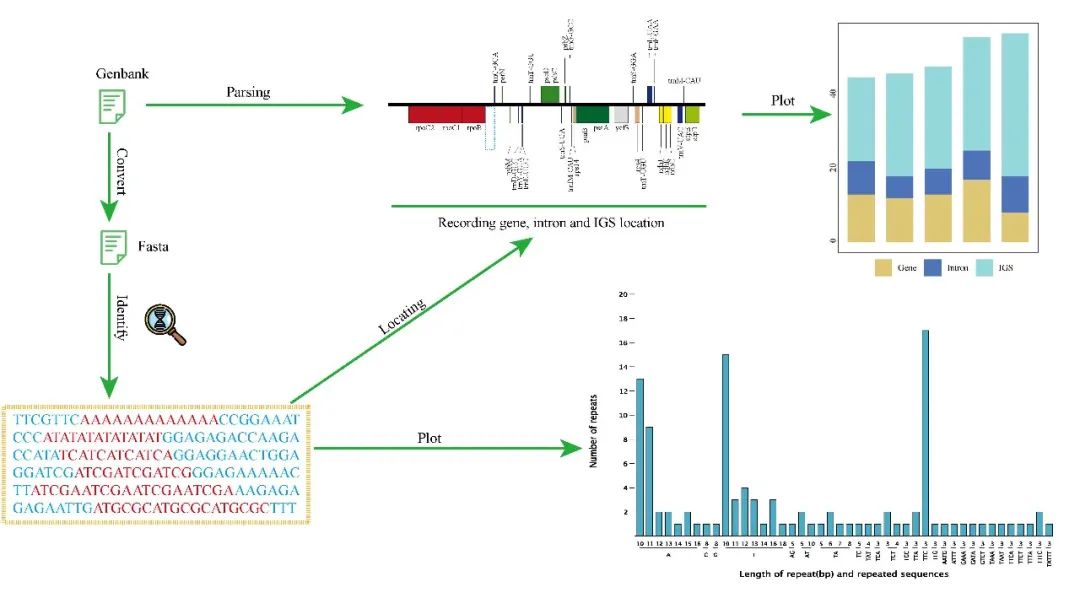
图4. CPStools中简单重复序列分析流程
目前,叶绿体基因组系统发育分析主要基于三种数据进行构建: 全叶绿体基因组、共有CDS序列和共有蛋白序列。“Seq”子命令能够高效获取共线性的全叶绿体基因组序列。CPStools中“phy”子命令含有两种提取模式:“CDS”和“Pro”,可分别提取和合并共有CDS和蛋白序列,为系统发育分析做数据准备。同时CPStools还包含三种格式转换模式,可将Genbank格式文件分别转换成Fasta、mVISTA和tbl格式。mVISTA格式文件可用于叶绿体基因组同源片段分析,tbl格式文件是后期数据上传NCBI所必须的文件。
讨 论
CPStools作为一个用户友好型软件,能够快速高效地进行叶绿体基因组数据分析,在保证分析准确性的同时,也显著提高了效率。但CPStools依赖Biopython解析GenBank和Fasta格式文件,因此这些文件必须严格遵循标准格式规范。我们推荐使用CPGAVAS2进行叶绿体基因组注释,其他软件的注释结果可能由于格式不匹配而与CPStools不兼容。研究人员在使用CPStools时应确保数据的兼容性。
代码和数据可用性
该软件包现已在PyPi (https://pypi.org/project/cpstools/) 和Conda (https://anaconda.org/bioconda/cpstools) 发布。文中所用的所有数据集也已上传至GitHub (https://github.com/Xwb7533/CPStools/tree/main/test_data)。在每个数据集中,我们都提供了详细的使用方法。同时我们也录制了该软件的视频教程 (https://www.bilibili.com/video/BV1fZ421K7nw/)。所有的补充材料(文本、图、表、中文翻译版本或视频)也可从线上获取。
引文格式:
Lijin Huang, Huanxi Yu, Zhi Wang, Wenbo Xu. 2024. CPStools: a package for analyzing chloroplast genome sequences. iMetaOmics e25. https://doi.org/10.1002/imo2.25
作者简介

黄丽瑾(第一作者)
● 中国药科大学博士研究生。
● 研究方向为药用植物次生代谢产物的生物合成与转录调控,相关学术成果已发表于iMetaOmics、Journal of experimental botany、International journal of molecular sciences等期刊。

于换喜(第一作者)
● 四川大学植物学硕士研究生,现就职于生态环境部南京环境科学研究所,工程师。
● 研究方向为植物分类学和保护生物学,相关学术成果已发表在iMetaOmics、Phytotaxa、AoB PLANTS、Frontiers in Plant Science等期刊。

徐文波( 通讯作者)
● 中国药科大学中药学硕士研究生。
● 研究方向为中药分子鉴定与细胞器基因组分析。已完成2000多种植物叶绿体基因组数据分析和百余种中药分子鉴定。相关学术成果已发表在iMetaOmics、Food Chemistry、Tree Physiology、Gene等期刊。
iMetaOmics
更多资讯
● iMeta姊妹刊iMetaOmics(定位IF>10)欢迎投稿!(2024.2.27)
● iMeta姊妹刊iMetaOmics编委招募 (定位IF>10) (2024.3.2)
● iMeta姊妹刊iMetaOmics电子版和印刷版ISSN申请获批(2024.4.1)
● iMeta姊妹刊iMetaOmics投稿系统正式上线(2024.4.17)
● iMeta姊妹刊iMetaOmics主编正式官宣(2024.4.22)
● 出版社iMetaOmics主页正式上线!(2024.4.28)
● iMetaOmics | 浙江大学宗鑫组揭示两猪种宿主-肠道菌群互作差异
● iMetaOmics | 罗鹏/袁硕峰/苗凯/程全发表STAGER: 生成式人工智能可靠性的标准化测试和评估推荐
● iMetaOmics | 徐州医科大杨欢组揭秘沙门氏菌-宿主-微生物群在免疫与代谢中的相互作
● iMetaOmics | 中科院动物所金坚石组综述16S rRNA基因扩增子测序技术的“前世今生”
● iMetaOmics | 浙大张天真组完成二倍体棉种泛基因组构建
● iMetaOmics | 张勇/李福平-先进糖蛋白组学在男性生殖研究中的潜在应用
● iMetaOmics | 暨南大学潘永勤/杨华组-炎症蛋白联合检测利于诊断甲状腺乳头状癌和结节性甲状腺肿
● iMetaOmics | 张开春组利用多组学方法揭示甜樱桃加倍后果色变化的候选基因
● iMetaOmics | 杜娟/林婷婷-慢性泪囊炎患者眼部菌群类型和纵向菌群变化
● iMetaOmics | 陈汉清/陈俊综述有关肝细胞癌治疗的新兴纳米医学策略
● iMetaOmics | 基因组所刘永鑫/卢洪评述微生物在提高杂种优势中的作用
● iMetaOmics | 上科大刘雪松组开发基于通路的肿瘤细胞鉴别工具TCfinder
● iMetaOmics | 中山大学刘鹏/邹宇田-整合人工智能实现HER2阳性乳腺癌精准管理
● iMetaOmics | 安徽农大李晓玉组-丛枝菌根真菌对玉米内生菌群的影响
● iMetaOmics | 徐涛/黄蓉/苏国海-急性冠脉综合征纵向多组学队列建设
● iMetaOmics | 通过整合宏组学促进人类与环境健康发展
●iMetaOmics | 苏州大学林俊组-揭示活性微生物及益生元/益生菌与关节炎联系
更多推荐
(▼ 点击跳转)
iMeta | 引用13000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

2卷2期封底

2卷4期封底

3卷2期

3卷3期

3卷3期封底

3卷4期

3卷4期封底

1卷1期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百千华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!发行后相继被Google Scholar、ESCI、PubMed、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.7,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,同学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,是定位IF>10的高水平综合期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science

腾讯云面向开发者汇聚海量精品云计算使用和开发经验,营造开放的云计算技术生态圈。
更多推荐



 1
1 0
0- 0



 已为社区贡献34条内容
已为社区贡献34条内容

所有评论(0)