
水稻(Oryza sativa)如何做GO富集分析
该脚本是第一个我用R写的小程序,虽然距离发布第一次用python写的小程序用于qRT-PCR结果计算的这篇推送相差一个月(https://mp.weixin.qq.com/s/HJFE4L0lQRJrGITCfl2UJg),但是阅读量却已经突破1w+,非常的意外。另外本公众号开通也有半年时间了,关注本公众号的人数也快突破1000。自己写的小程序拿出来免费供全网参考使用,在这过程中我也收到了许许多多
在RNA-seq数据的分析中,非常普遍的做法是先筛选出差异表达基因,然后针对筛选出来的差异基因做GO富集分析,以了解差异表达基因的功能。虽然说测序公司在最后的报告中会给出关于差异表达基因的GO富集结果,但是公司给的结果是流程化的,不够个性化,大多时候需要自己筛选感兴趣的基因集重新进行GO富集分析。关于如何从大量的基因中筛选到自己感兴趣的基因集,可以参考《转录组测序(RNA-seq)的数据如何分析?》这一篇文章(https://mp.weixin.qq.com/s/FOURrp-ca6X72BZzqoXHCA)。目前虽然有一些在线网站可以进行富集分析,但是有些可能需要付费或者不能批量进行GO富集分析,因此用R写了一个可以批量进行GO富集分析的小程序。
1.R包的下载与获取
1.1下载tidyverse, openxlsx, clusterProfiler三个R包
install.packages('openxlsx') #用于excel数据的读取
install.packages('tidyverse') #用于数据的处理和作图
install.packages('argparser') #用于命令行参数的解析
BiocManager::install('clusterProfiler') #用于数据的读取
1.2下载org.Osativa.eg.db包
由于在Bioconductor下还没有水稻的OrgDb包,因此我根据MSU数据库[1]的水稻注释自己构建了一个水稻的OrgDb,大家只需要直接下载放到R的library目录下(新手推荐这种方式,后续也不用修改我写的程序代码),或者下载到任意一个文件夹(图1),但在加载R包的时候指定一下R包的目录即可,修改我后面R文件的library的部分的代码,需要指定目录加载R包。下载地址:
链接: https://pan.baidu.com/s/1Q1OQeXn1nIXIWSGuvI0HRw
提取码: ifgh
#指定目录加载R包,lib = 'R包的路径'
library(org.Osativa.eg.db, lib = 'D:\\浙江大学实验\\Oryza sativa genome\\Osativa_enrichment\\R_library')

图1 org.Osativa.eg.db的文件夹位置
2.读取用于GO富集分析的基因集(EXCEL文件)
首先建立一个名为original_file的文件夹(图2)(注意文件夹名称一定要是original_file,不然程序运行不了)。

图2 建立original_file的文件夹
然后进入名为"original_file"的文件夹,把差异表达的基因列表放到excel文件中,注意excel文件的基因ID一要是要是“LOC_Os02g43410”这种形式的基因ID,不能是Os开头的基因ID,如是Os开头的则需要进行转化。在后续我也会更新一个LOC和Os两个进行基因ID批量转化的小程序,敬请期待!然后基因ID这一列的行名一定要是“gene_id”,不然程序识别不了那列是基因ID(图3)。

图3 用于GO富集分析的excel表格的样式
类似这样的用于富集分析的excel文件只要基因ID这列名称为“gene_id”,无论有多少个文件,都可快速的进行富集分析,主要把文件都放在"original_file"的文件夹下即可。例如我一次性要进行3个基因集的GO富集分析,那就把三个基因集分别放到3个excel文件中(图4),每个excel文件只放一个名称为“gene_id”的基因集合。
(PS:excel文件的名称决定了后续的结果文件的名称。例如我的excel文件时first,那么最后生成的结果也是命名成为first的excel文件和pdf图片)

图4 "original_file"文件夹下的样式
3.建立名称为“table”和“plot”文件夹
在跟“original_file”文件夹同级的目录下,建立名称分别为“table”和“plot”的两个文件夹用于GO富集结果的存储。一定要建立这两个文件夹,且名称要一样,否则程序运行不了(图5)。

图5 建立名称为“table”和“plot”文件夹
4.下载Osativa_GO_enrichment.R
将一下代码复制到名称为“Osativa_GO_enrichment.R”的文件中。可以先建立一个txt文件,然后把下面的代码复制进入保存后重命名为“Osativa_GO_enrichment.R”,也可以直接下载Osativa_GO_enrichment.R文件,下载地址见最后的附件。注意“Osativa_GO_enrichment.R”文件也要与“original_file”、“table”和“plot”同级(图6)。

图6 “Osativa_GO_enrichment.R”文件的位置
“Osativa_GO_enrichment.R”文件的代码内容如下:
library(tidyverse)
library(openxlsx)
library(org.Osativa.eg.db)
library(clusterProfiler)
library(argparser, quietly=TRUE)
# 创建参数解析对象
p <- arg_parser("GO enrichment analysis in rice")
# Add command line arguments
p <- add_argument(p, "path", help="input: gene ID filepath", type="character" )
# Parse the command line arguments
argv <- parse_args(p)
path <- argv$path
setwd(path)
path_list <- list.files(path = "./original_file/") %>%
str_remove(pattern = '.xlsx')
for (i in path_list) {
#读取基因集文件
gene_id <- read.xlsx(sprintf('./original_file/%s.xlsx', i)) %>%
pull(gene_id)
#进行GO富集分析
de_ego <- enrichGO(gene_id,
OrgDb = org.Osativa.eg.db,
keyType = 'GID',
ont = 'ALL',
qvalueCutoff = 1,
pvalueCutoff = 1)
df_ego <- as.data.frame(de_ego)
#将富集结果存入excel表格中
write.xlsx(df_ego, file = sprintf('./table/%s_table.xlsx', i))
#将GeneRatio中的分数转化为小数
df_ego$GeneRatio <- apply(str_split(df_ego$GeneRatio, "/", simplify = T), 2,as.numeric)
df_ego$GeneRatio <- df_ego$GeneRatio[,1]/df_ego$GeneRatio[,2]
#按照pvalue值进行排序,生成Top10
df_ego <- arrange(df_ego, Count) %>%
group_by(ONTOLOGY) %>%
top_n(-10, wt=pvalue) %>%
mutate(Description = substr(Description, 1, 50)) %>%
arrange(Count) %>%
distinct(Description, .keep_all = T) %>%
mutate(Description = factor(Description, levels = Description))
#用ggplot2对结果进行可视化
pdf(sprintf('./plot/%s_plot.pdf', i), width = 6, height = 8)
p <- ggplot(data = df_ego, aes(x = GeneRatio, y = Description))+
geom_point(shape = 21, aes(fill = p.adjust, size = Count)) +
scale_fill_gradient(low = 'red', high = 'purple')+
facet_grid(factor(ONTOLOGY)~.,
scales = 'free')+
theme_bw()+
labs(x = "GeneRatio")
print(p)
dev.off()
}
print("\n")
print("\n")
print("\n")
print("\n")
print("\n")
print("########################## GO enrichment analysis have finished #########################")
5.在终端(Terminal)运行R脚本
运行的脚本如下:
Rscript "D:\生物信息学习\GO可视化\Osativa_GO_enrichment.R" "D:\生物信息学习\GO可视化"
运行的R脚本主要包括3部分,一个是Rscript,然后空格输入Osativa_GO_enrichment.R文件的绝对路径,在空格,输入自己的工作路径,即“original_file”、“table”和“plot”文件的当前目录的路径(图7和图8),这三部分输入完成后,按一个回车即可(运行时关闭所有的excel文件)。

图7 命名行的3部分
图8 “original_file”、“table”和“plot”文件的当前路径
出现如下显示则表示运行成功。
可以进入table和plot文件夹进行查看结果文件,如下图所示。



6.GO富集结果准确性的验证
为了验证GO富集结果的准确性,特意挑了一篇发表在nature communications上的一篇文章[2]进行比较,左边(图9a)为我们做出来的GO富集结果,右边(图9b)为nature communications的文章里面的富集结果。比较后发现绝大部分GO terms时一致的,有少数term的描述稍有不同,但内容一致。因此结果可靠。
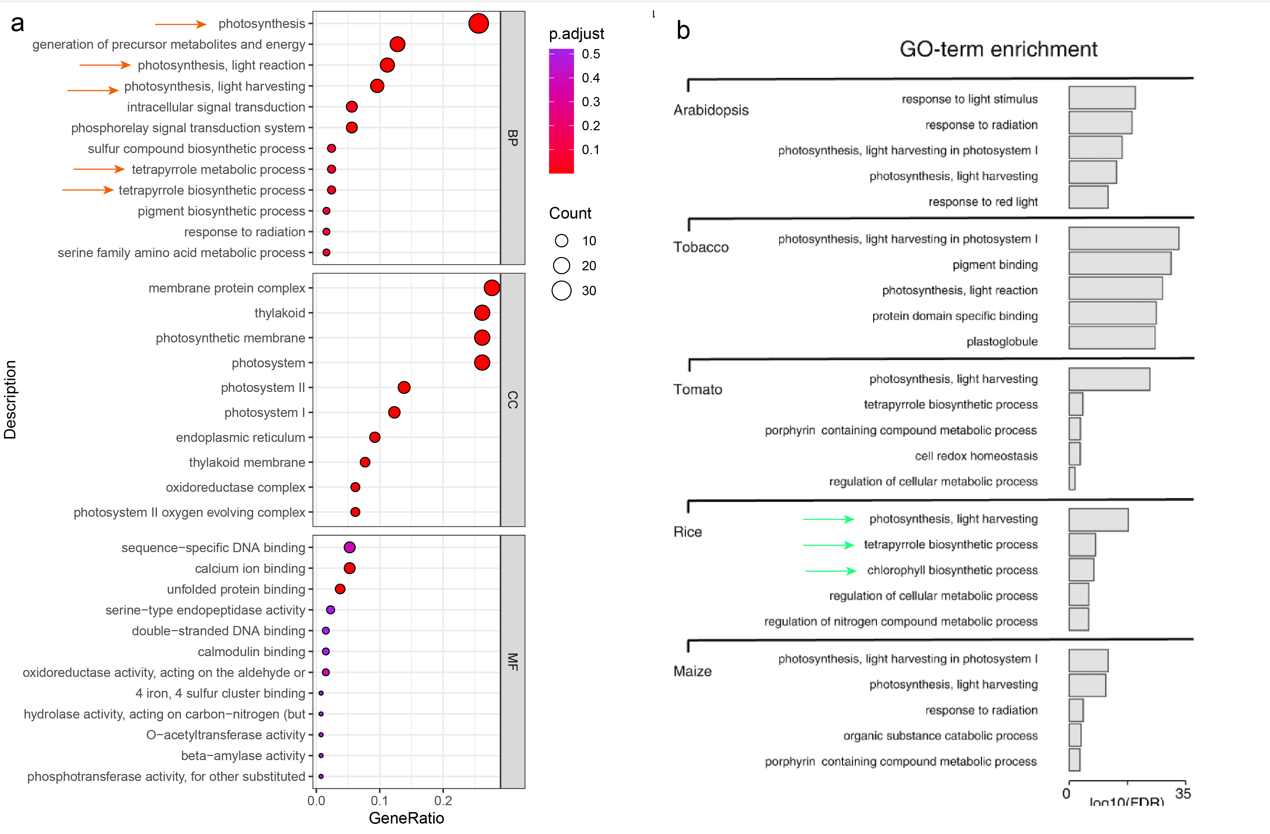
图9 GO富集结果的比较
写在最后
该脚本是第一个我用R写的小程序,虽然距离发布第一次用python写的小程序用于qRT-PCR结果计算的这篇推送相差一个月(https://mp.weixin.qq.com/s/HJFE4L0lQRJrGITCfl2UJg),但是阅读量却已经突破1w+ ,非常的意外。另外本公众号开通也有半年时间了,关注本公众号的人数也快突破1000。

今天想起我当初开通此公众号的主要目的只是用于自己的生信学习,是当作笔记来做的,方便以后自己用到时可以查看,还有好多地方写得不够完善,但却得到了这么多人的关注,对此我非常感谢大家的支持。
自己写的小程序拿出来免费供全网参考使用,在这过程中我也收到了许许多多来自于素未相识的人的一些反馈,这些反馈又使我进一步优化了自己的小程序。我想这就是这就是开源精神的强大之处吧,也是计算机发展的如此快的重要因素之一吧。
参考文献
[1] Kawahara, Y., de la Bastide, M., Hamilton J. P., Kanamori, H., McCombie, W. R., Ouyang, S., Schwartz, D. C., Tanaka, T., Wu, J., Zhou, S., Childs, K. L., Davidson, R. M., Lin, H., Quesada-Ocampo, L., Vaillancourt, B., Sakai, H., Lee, S. S., Kim, J., Numa, H., Itoh, T., Buell, C. R., Matsumoto, T. 2013. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6:4.
[2] Tu X, Ren S, Shen W, Li J, Li Y, Li C, Li Y, Zong Z, Xie W, Grierson D, Fei Z, Giovannoni J, Li P, Zhong S (2022) Limited conservation in cross-species comparison of GLK transcription factor binding suggested wide-spread cistrome divergence. Nature communications 13 (1):7632. doi:10.1038/s41467-022-35438-4
附件
链接: https://pan.baidu.com/s/1vQkCmrBIHrxWuV10Yl-u_Q
提取码: qc1q

旨在为数千万中国开发者提供一个无缝且高效的云端环境,以支持学习、使用和贡献开源项目。
更多推荐



 4
4 0
0- 0



 已为社区贡献2条内容
已为社区贡献2条内容

所有评论(0)