Rliger | 完美整合单细胞测序数据(部分交集数据的整合)(三)
11. 写在前面对于只有只有部分重叠的datasets,合并方法我们依然可以采用Seurat、Harmony,rliger包,本期介绍一下rliger包的用法。22.用到的包rm(list = ls())library(Seurat)library(SeuratDisk)library(SeuratWrappers)library(patchwork)library(harmony)library

11. 写在前面
对于只有只有部分重叠的datasets,合并方法我们依然可以采用Seurat、Harmony,rliger包,本期介绍一下rliger包的用法。
22.用到的包
rm(list = ls())
library(Seurat)
library(SeuratDisk)
library(SeuratWrappers)
library(patchwork)
library(harmony)
library(rliger)
library(RColorBrewer)
library(tidyverse)
library(reshape2)
library(ggsci)
library(ggstatsplot)
33. 示例数据
这里我们提供1个3’ PBMC dataset和1个whole blood dataset。🤗
umi_gz <- gzfile("./GSE149938_umi_matrix.csv.gz",'rt')
umi <- read.csv(umi_gz,check.names = F,quote = "")
matrix_3p <- Read10X_h5("./3p_pbmc10k_filt.h5",use.names = T)
创建Seurat对象。 🧐
srat_wb <- CreateSeuratObject(t(umi),project = "whole_blood")
srat_3p <- CreateSeuratObject(matrix_3p,project = "pbmc10k_3p")
rm(umi_gz)
rm(umi)
rm(matrix_3p)
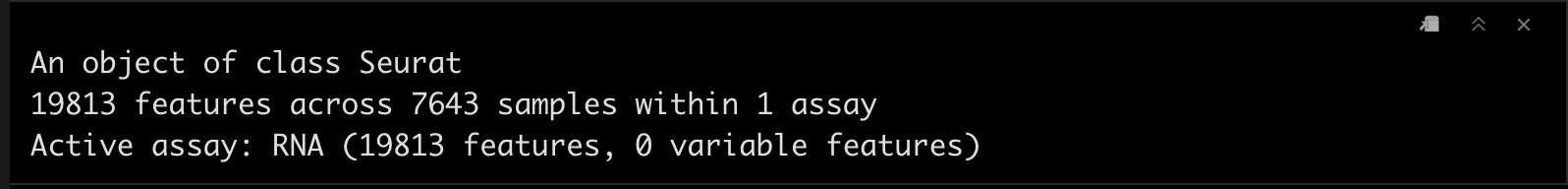
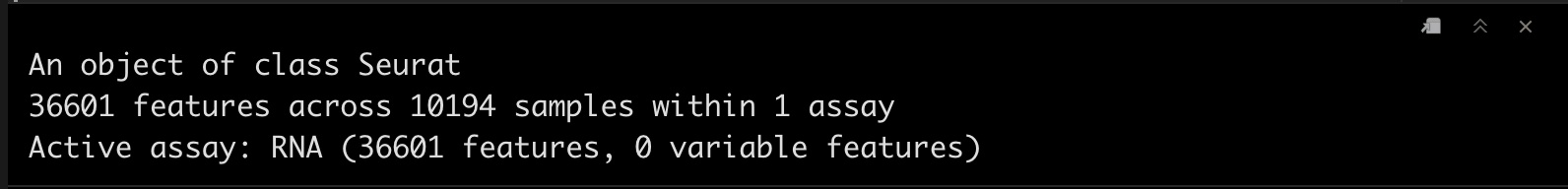
srat_wb
srat_3p
44. 修改metadata
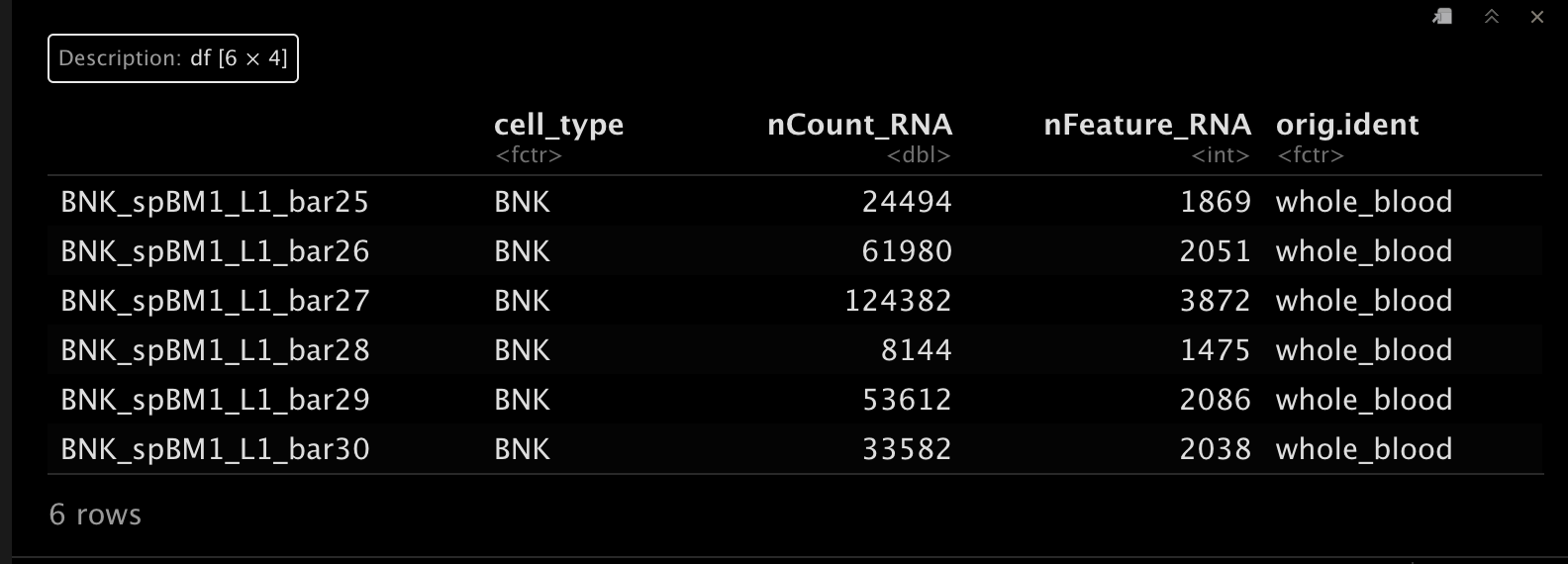
为了方便后续分析,这里我们对metadata进行一下注释修改。
colnames(srat_wb@meta.data)[1] <- "cell_type"
srat_wb@meta.data$orig.ident <- "whole_blood"
srat_wb@meta.data$orig.ident <- as.factor(srat_wb@meta.data$orig.ident)
head(srat_wb[[]])
55. 初步合并
5.1 简单合并
这里我们先用merge将2个数据集简单合并在一起。(这里我们默认做过初步过滤了哈,具体的大家可以看一下之前的教学。)😘
wb_liger <- merge(srat_3p,srat_wb)
5.2 标准操作
我们在这里做一下Normalization,寻找高变基因等等标准操作。👀 Note! 这里需要跟大家说下,rlinger在ScaleData时没有将数据中心化,我们需要设置为F。
wb_liger <- NormalizeData(wb_liger)
wb_liger <- FindVariableFeatures(wb_liger)
wb_liger <- ScaleData(wb_liger, split.by = "orig.ident", do.center = F)
5.3 合并数据
wb_liger <- RunOptimizeALS(wb_liger, k = 30, lambda = 5, split.by = "orig.ident")
wb_liger <- RunQuantileNorm(wb_liger, split.by = "orig.ident")
66. 降维与聚类
6.1 寻找clusters
wb_liger <- FindNeighbors(wb_liger,reduction = "iNMF",k.param = 10,dims = 1:30)
wb_liger <- FindClusters(wb_liger)
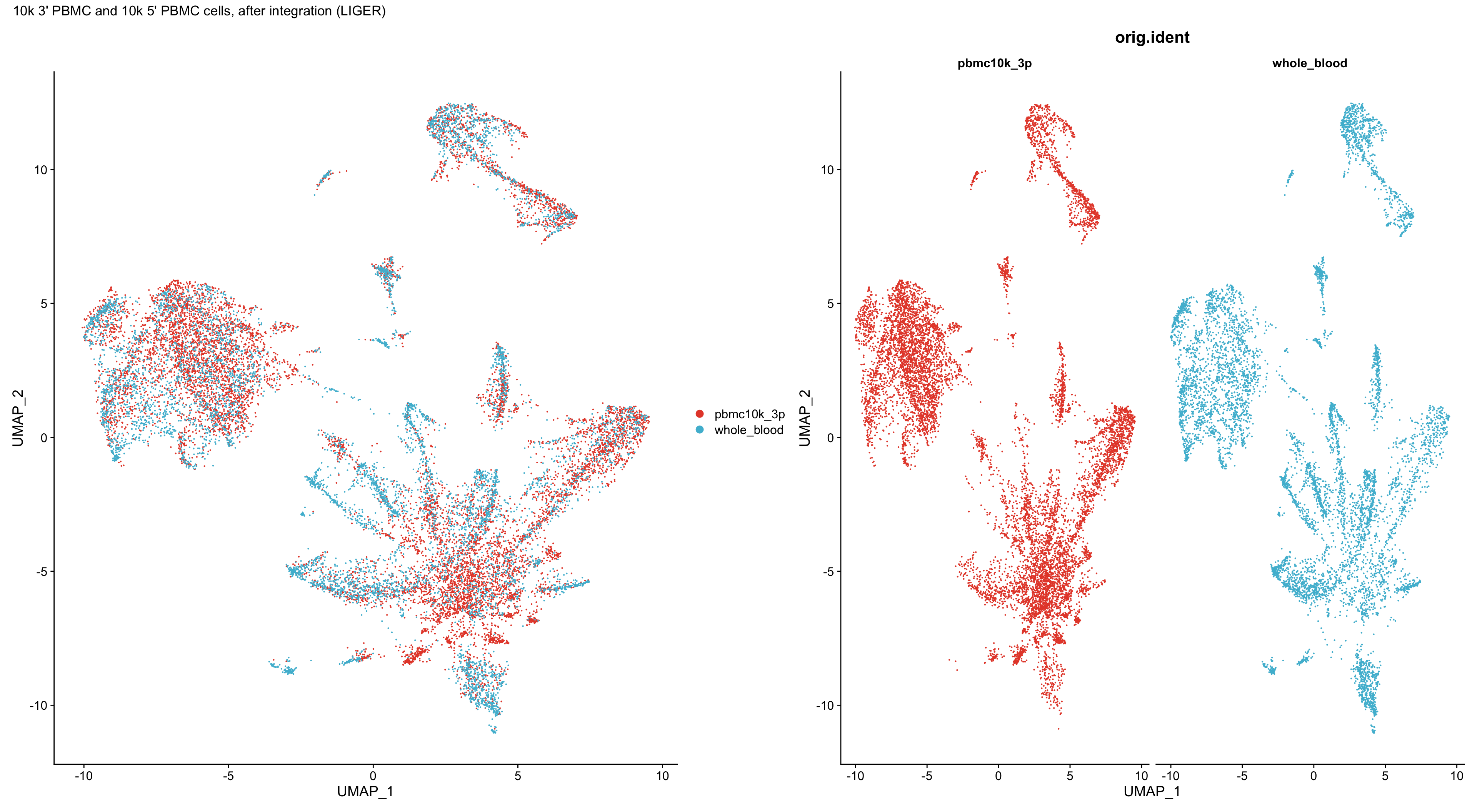
6.2 聚类可视化
wb_liger <- RunUMAP(wb_liger, dims = 1:ncol(wb_liger[["iNMF"]]),
reduction = "iNMF",verbose = F)
wb_liger <- SetIdent(wb_liger,value = "orig.ident")
p1 <- DimPlot(wb_liger,reduction = "umap") +
scale_color_npg()+
plot_annotation(title = "10k 3' PBMC and 10k 5' PBMC cells, after integration (LIGER)")
p2 <- DimPlot(wb_liger, reduction = "umap",
group.by = "orig.ident", pt.size = .1, split.by = 'orig.ident') +
scale_color_npg()+
NoLegend()
p1 + p2
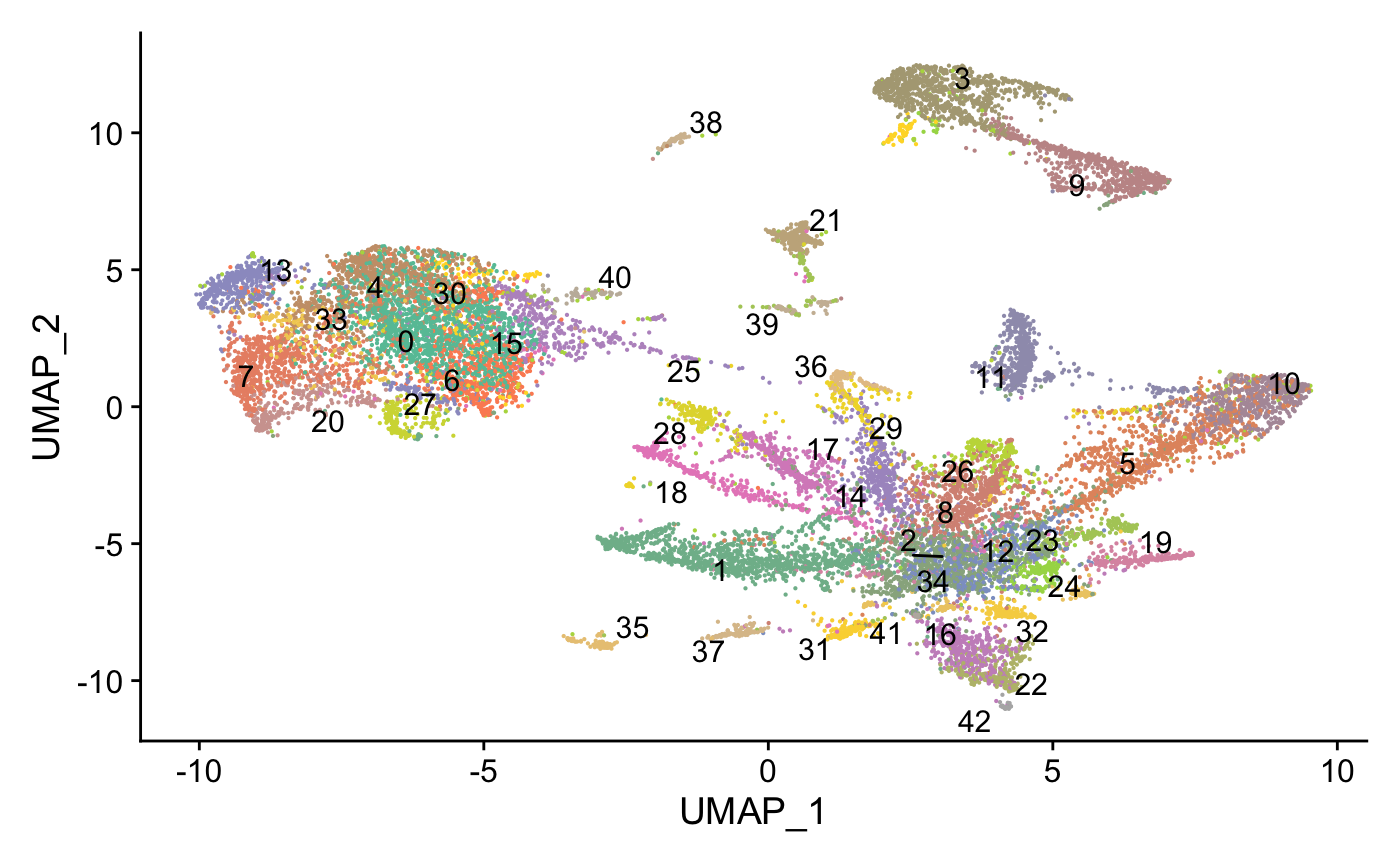
wb_liger <- SetIdent(wb_liger,value = "seurat_clusters")
ncluster <- length(unique(wb_liger[[]]$seurat_clusters))
mycol <- colorRampPalette(brewer.pal(8, "Set2"))(ncluster)
DimPlot(wb_liger,reduction = "umap",label = T,
cols = mycol, repel = T) +
NoLegend()
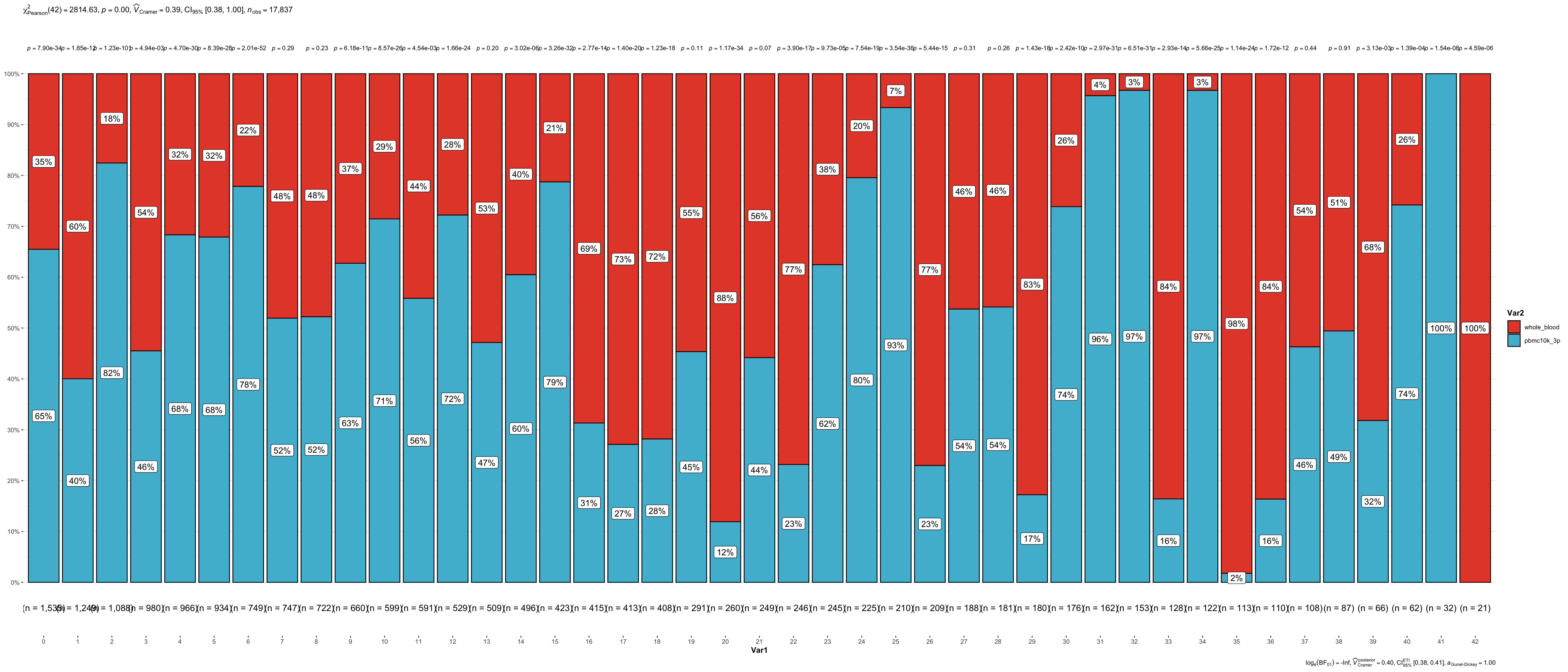
6.3 具体查看及可视化
我们看下各个clusters在两个datasets各有多少细胞。
count_table <- table(wb_liger@meta.data$seurat_clusters, wb_liger@meta.data$orig.ident)
count_table
#### 可视化
count_table %>%
as.data.frame() %>%
ggbarstats(x = Var2,
y = Var1,
counts = Freq)+
scale_fill_npg()

需要示例数据的小伙伴,在公众号回复
Merge2获取吧!点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰
📍 🤩 ComplexHeatmap | 颜狗写的高颜值热图代码!
📍 🤥 ComplexHeatmap | 你的热图注释还挤在一起看不清吗!?
📍 🤨 Google | 谷歌翻译崩了我们怎么办!?(附完美解决方案)
📍 🤩 scRNA-seq | 吐血整理的单细胞入门教程
📍 🤣 NetworkD3 | 让我们一起画个动态的桑基图吧~
📍 🤩 RColorBrewer | 再多的配色也能轻松搞定!~
📍 🧐 rms | 批量完成你的线性回归
📍 🤩 CMplot | 完美复刻Nature上的曼哈顿图
📍 🤠 Network | 高颜值动态网络可视化工具
📍 🤗 boxjitter | 完美复刻Nature上的高颜值统计图
📍 🤫 linkET | 完美解决ggcor安装失败方案(附教程)
📍 ......

本文由 mdnice 多平台发布

为开发者提供学习成长、分享交流、生态实践、资源工具等服务,帮助开发者快速成长。
更多推荐



 1
1 0
0- 0



 已为社区贡献4条内容
已为社区贡献4条内容

所有评论(0)