amber的安装和使用
目录一、基础环境二、安装ambertools本文参考:https://blog.csdn.net/qq_33953882/article/details/113995531https://drugai.blog.csdn.net/article/details/103456040Amber18安装(非root用户)_Code Wang的博客-CSDN博客_amber18安装一、基础环境1.初始环境
之目录
本文参考:
https://blog.csdn.net/qq_33953882/article/details/113995531
https://drugai.blog.csdn.net/article/details/103456040
Amber18安装(非root用户)_Code Wang的博客-CSDN博客_amber18安装
一、基础环境
1.初始环境情况
- Ubuntu 18.04.5 LTS (GNU/Linux 5.4.0-81-generic x86_64)
- cmake version 3.5.1
- gcc (Ubuntu 5.4.0-6ubuntu1~16.04.12) 5.4.0 20160609
- GeForce GTX 1080*8
- Driver Version: 460.84
- CUDA Version: 11.2

2. 依赖库的安装
apt -y update
apt -y install tcsh make
apt -y install gcc gfortran
apt -y install flex bison patch
apt -y install bc xorg-dev libbz2-dev wget
有条件这些依赖库尽量都安装,否则后面的安装中可能会出现下图报错,类似这种问题就是因为你有依赖库没装,具体是哪个我忘记了:

3. 安装conda和cuda
1. miniconda
因为miniconda安装比较方便快捷,所以选择了安装miniconda。参考了https://blog.csdn.net/weixin_42066885/article/details/80323173。
首先进入miniconda资源网址https://docs.conda.io/en/latest/miniconda.html下载对应资源,我选择的是这个

之后上传服务器,进入文件夹安装
bash Miniconda3-latest-Linux-x86_64.sh会有一大堆回车,让你看完一个license,之后需要手动键入一个yes

之后一直回车就行,后来问你是否要初始化,我选择的是yes

之后用source命令激活环境
source ~/.bashrc
就可以进入base环境。

2. cuda
amber18和ambertool19只能支持10.2以下的cuda版本。可以不用root权限仅在自己的user上装一个10.2,详见:Linux服务器下给当前用户安装自己的CUDA、CUDA 还是自己的好用 【有效安装教程】_墨理学AI的博客-CSDN博客_服务器安装cuda
cuda最新版的网址为https://developer.nvidia.com/cuda-downloads,但这上面最低只有ubuntu 18,我用的服务器是16的,后来找到了这个网址:
https://developer.nvidia.com/cuda-toolkit-archive在这里可以下载16和17。因为有blog里说amber18最高支持cuda 9.2,所以我就选了9.0。

3. 安装anaconda
从清华源镜像下载合适镜像(我选择用最新版)

上传到服务器上后执行
sh Anaconda3-5.3.1-Linux-x86_64.sh回车所有,遇见让输入yes的就输入yes,最后这里输入no
之后配置环境变量:
vim ~/.bashrc在最后加一句(username是你自己的用户名)
export PATH="/home/username/anaconda3/bin:$PATH"
之后执行
source ~/.bashrc就可以正常使用了。可以用 which python 查看python解释器位置检查是否配置好环境变量。或用conda --version 检查conda是否安装成功。
4. 创建环境
这里不知道是为什么,按照上述配置好之后,第一次用conda activate base会无效
就很尴尬,必须先用source activate base,激活之后conda 的命令就可以正常执行了。
这样很麻烦,可能是新版conda有点改动。根据提醒把~/.bashrc中形如:
![]()
去掉,再改成:
![]()
就可以正常使用conda命令了。
可以创建环境
conda create -n myname python=3.7创建成功之后可以查看环境列表
conda env list
5. 使用命令记录
退出环境
conda deactivate
删除环境
conda remove -n myname --all二、安装Amber
1. 下载后上传服务器并解压
tar jxvf Amber18.tar.bz2
tar jxvf AmberTools19.tar.bz2
注意,这里我用的是18版本,AmberTools虽然是19,但解压出来依然是18。解压之后应该会只有一个amber18的文件夹

这与新版是不同的,新版的AmberTools21解压出来会是一个amber20和一个amber20_src两个文件夹,21版执行安装也不同。21版仅AmberTools安装如下:
1. 进入文件夹
cd amber20_src/build2. 编译
./run_cmake3. 执行安装
make install
因为我要用的是18版,所以以下只记录18版安装。
2. 配置环境变量
上一节也介绍过,需要安装一些环境依赖(再做一次提醒)
apt-get install bison bc csh flex gfortran g++ xorg-dev zlib1g-dev libbz2-dev patch python环境变量配置:
vi ~/.bashrc #打开 bashrc文件
#Amber18
test -f /opt/amber18/amber.sh && source /opt/amber18/amber.sh
export AMBERHOME=/opt/amber18
export CUDA_HOME=/usr/local/cuda
export PATH=$PATH:$CUDA_HOME/bin
export LD_LIBRARY_PATH=$LD_LIBRARY_PATH:$CUDA_HOME/lib64
source ~/.bashrc #source 后立即生效注意上述 test 和 source 后的目录需要改成解压后的目录,比如我就是

3. 安装openmpi
这里是并行版本必须安装的一个包,用以下命令:
CC=gcc CXX=g++ FC=gfortran ./configure --prefix=/home/$USERNAME/openmpi4. 更新补丁(一定一定一定要更新)
因为第一次安装没有更新补丁,在装gpu并行版本时(cpu并行时也有类似问题,但cpu串行正常)出现这样一个bug:

如果只用 make install ,则报错如下

后来问了同门才发现这是因为有一些更新,若不更新会出问题。更新补丁用:
./update_amber --update另同门给了我他的安装命令记录。
下面安装的时候还出了个问题:
/usr/bin/ld: cannot find -lcublas解决方法参考这里/usr/bin/ld: cannot find -lcublas 问题的解决办法_qq_42550613的博客-CSDN博客
就是在你设定的cuda目录里找到他生成的libcublas.so 文件,放到/usr/local/cuda-9.1/lib64/libcublas.so(设定的cuda目录)这个里面。找这个.so文件可以用以下命令
locate libcublas.so5. 编译测试具体版本
编译cpu串行版本
cd $AMBERHOME
./configure --with-python /root/anaconda3/bin/python gnu
test -f /opt/amber/amber18/amber.sh && source /opt/amber/amber18/amber.sh
make install -j 32
# Testing
make test编译cpu并行版本
# 编译并行版pmemd:pmemd.MPI
cd $AMBERHOME
./configure --with-python /root/anaconda3/bin/python -mpi gnu
test -f /opt/amber/amber18/amber.sh && source /opt/amber/amber18/amber.sh
make install
# bashrc中添加
export DO_PARALLEL="mpirun --allow-run-as-root -np 4 "
# 编译并行版cpptraj和NAB AmberTools19 OpenMP
./configure --with-python /usr/bin/python -openmp gnu
test -f /opt/amber/amber18/amber.sh && source /opt/amber/amber18/amber.sh
make openmp编译gpu版本(要钱的,组里买了使用权)
#GPU版本:pmemd.cuda
cd $AMBERHOME
./configure --with-python /root/anaconda3/bin/python -cuda gnu
test -f /opt/amber/amber18/amber.sh && source /opt/amber/amber18/amber.sh
make install
#GPU并行版本 pmemd.cuda.MPI
./configure --with-python /root/anaconda3/bin/python -cuda -mpi gnu
test -f /opt/amber/amber18/amber.sh && source /opt/amber/amber18/amber.sh
make install
# Testing
make test.cuda
make test.cuda_parallel以下贴一些截图
没装之前:

![]()
装完以后:

安装过程部分截图:
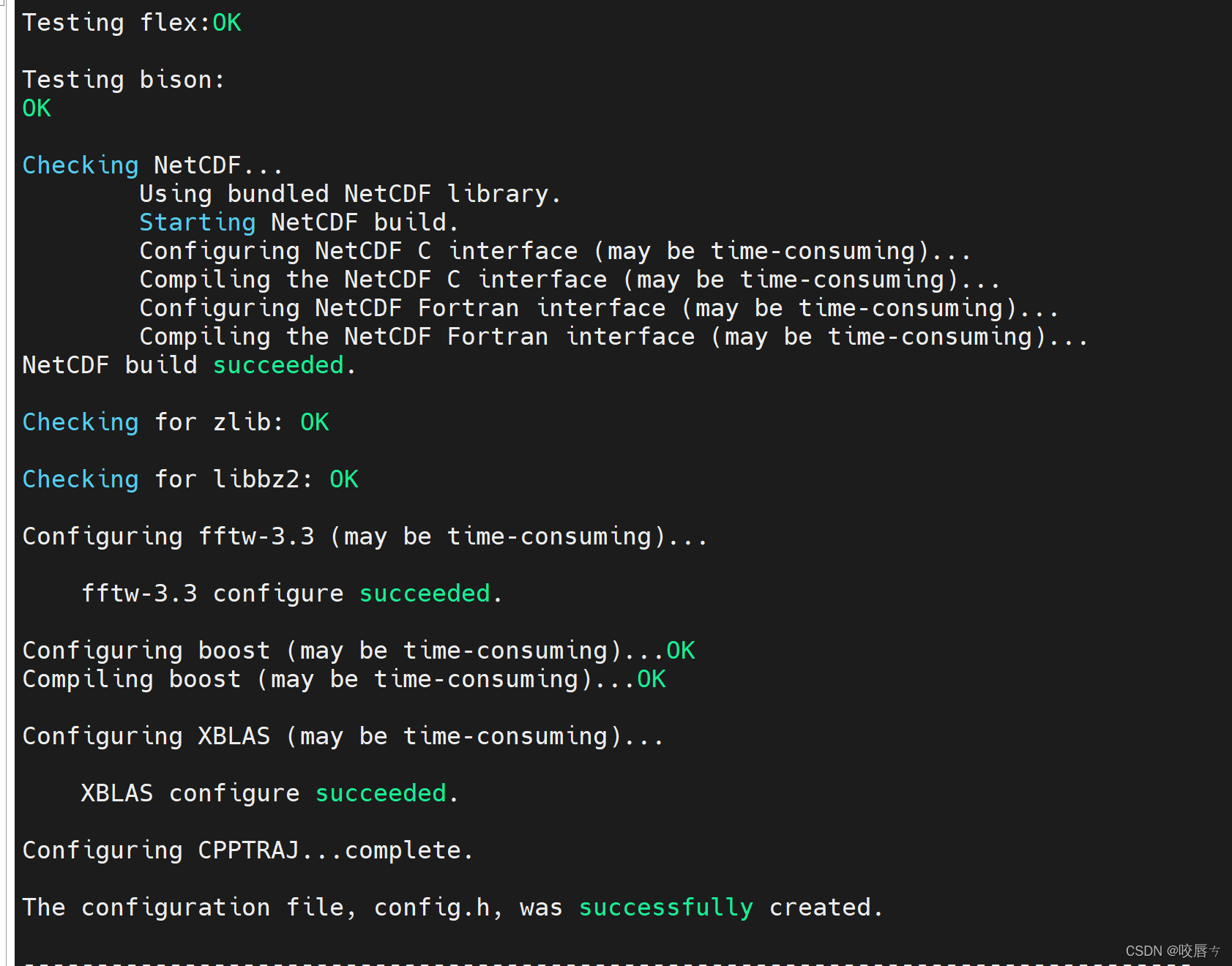
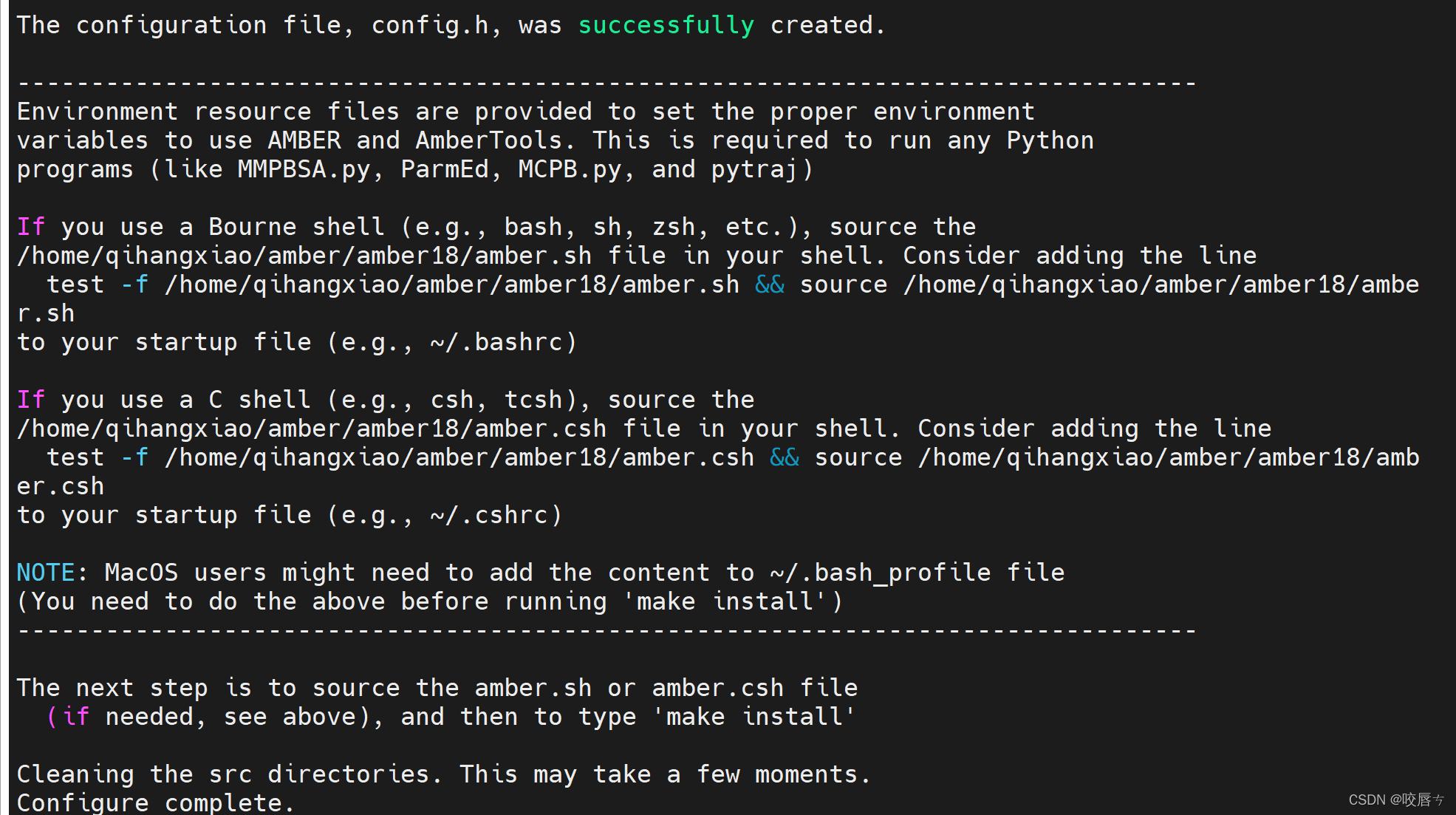
我需要用的是并行版pmemd,也就是pmemd.cuda命令,实验如下即为安装成功,填入必要参数则正常使用,参数需看官方文档:Amber Manuals,以及官方文档:Amber Tutorials

为开发者提供学习成长、分享交流、生态实践、资源工具等服务,帮助开发者快速成长。
更多推荐



 18
18 0
0- 0



 已为社区贡献2条内容
已为社区贡献2条内容

所有评论(0)