
分子对接及结果分析在线工具
简单介绍GRAMM,ZDOCK和PDBePISA分子对接及结果分析的流程。
分子对接(docking)有助于理解分子之间的相互作用。分子对接过程中,受体和配体是柔性的,即在结合过程中靶酶和底物的分子构象是变化的,不仅要满足空间的匹配,也要满足结合自由能的匹配。1958年D.E.Koshland提出了 “诱导契合"学说,核心内容为蛋白的活性位点通过与配体的相互作用而发生变化。实际上,“诱导契合"比“锁钥模型"更加准确。另一方面,计算机科学的发展进一步促成了分子对接的发展。1995年,Accelrys公司开发的计算化学软件Affinity上市,这是第一个可以进行大分子参与对接的软件,此后各种软件层出不穷。
分子对接是通过研究配体分子和受体分子的相互作用,预测其亲和力和结合模式,实现基于结构的药物设计和筛选的一种重要方法。配体与受体结合必须满足互相匹配原则,即配体与受体几何形状、静电、氢键、疏水相互作用互补匹配。
分子对接是将已知三维结构数据库中的分子逐一放在靶标分子的活性位点处,通过不断优化受体化合物的位置、构象、分子内部可旋转键的二面角和受体的氨基酸残基侧链和骨架,寻找受体小分子化合物与靶标大分子作用的最佳构象,并预测其结合模式、亲和力,通过打分函数挑选出接近天然构象的与受体亲 和力最佳的配体的过程。
(1) 刚性对接:刚性对接方法在计算过程中,参与对接的分子构像不发生变化,仅改变分子的空间位置与姿态,刚性对接方法的简化程度最高,计算量相对较小,适合于处理大分子之间的对接。
(2) 半柔性对接:半柔性对接方法允许对接过程中小分子构像发生一定程度的变化,但通常会固定大 分子的构像,另外小分子构像的调整也可能受到一定程度的限制,如固定某些非关键部位的键长、键角等, 半柔性对接方法兼顾计算量与模型的预测能力,是应用比较广泛的对接方法之一。
(3) 柔性对接:柔性对接方法在对接过程中允许研究体系的构像发生自由变化,由于变量随着体系的原子数呈几何级数增长,因此柔性对接方法的计算量非常大,消耗计算机时很多,适合精确考察分子间识别情况。
常用的分子对接软件有 AutoDock、DOCK、3D-DOCK、HEX、 HADDOCK、Z-DOCK、GRAXX 等,其中HADDOCK、Z-DOCK、GRAMM-X 等可以做大分子与大分子对接。下面以GRAMM-X为例。
GRAMM-X -分子对接
http://vakser.compbio.ku.edu/resources/gramm/grammx/
vakser工作室的软件。
此软件与原始GRAMM不同,不过两个程序包都使用FFT来全局搜索最佳刚体构象,所以GRAMM-X做的是刚性对接。(对接还有柔性对接和半柔性对接)
注意:此服务器将忽略输入文件中的任何小配体或其他非蛋白质分子。
它专为对接蛋白质分子对而设计。
受体配体填入顺序其实随意,计算原理是受体不动配体在受体表面滚动找到结合位点。
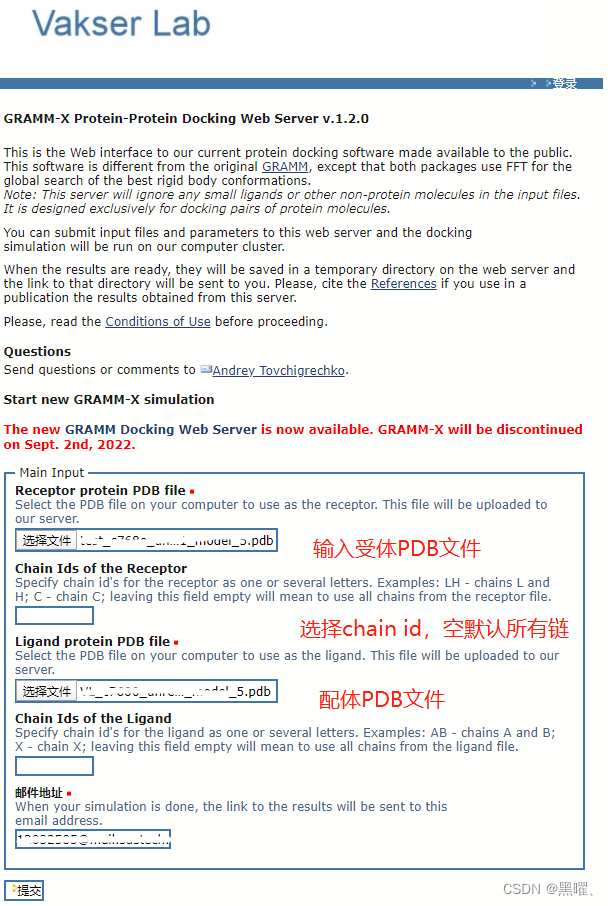
输出参数可以按默认值提交,只是计算时间长一点。
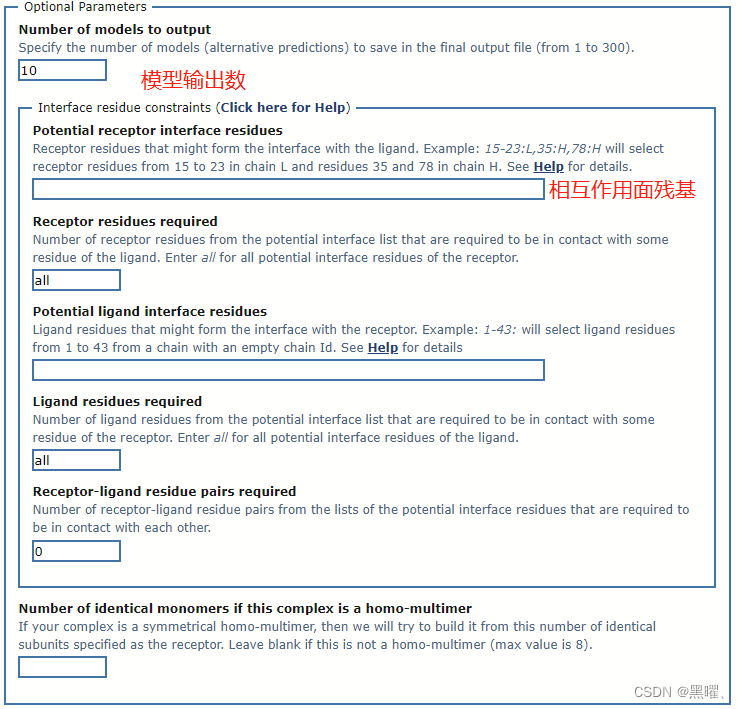
结果将保存在 Web 服务器上的一个临时目录中,个人邮箱会收到该目录的链接。预测结果是一个pdb文件。
注意页面有一段红字:新的RAMM对接Web服务器现已面市。GRAMM-X将于2022年9月2日停产。
所以试试新网站:
http://gramm.compbio.ku.edu/
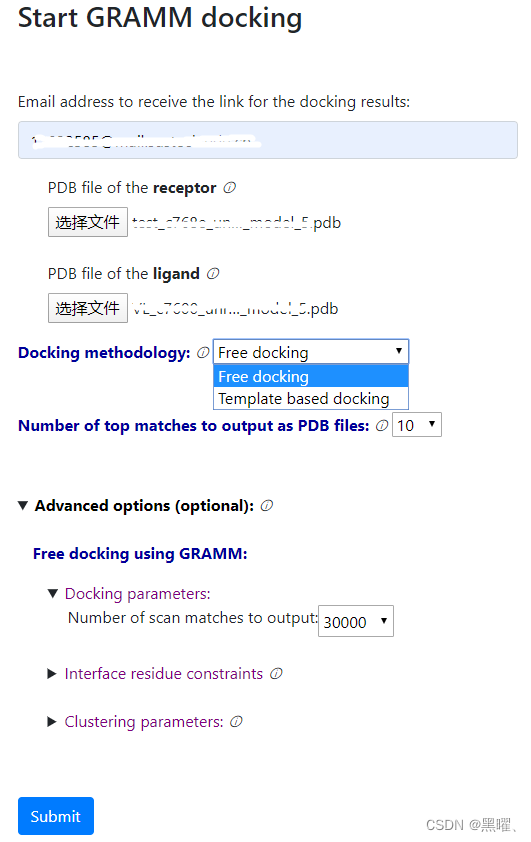
参数基本还是那些,界面更简洁漂亮了。
过大概半小时左右会收到邮件,进入结果链接,点击PDB file下载文件(GRAMM-X):
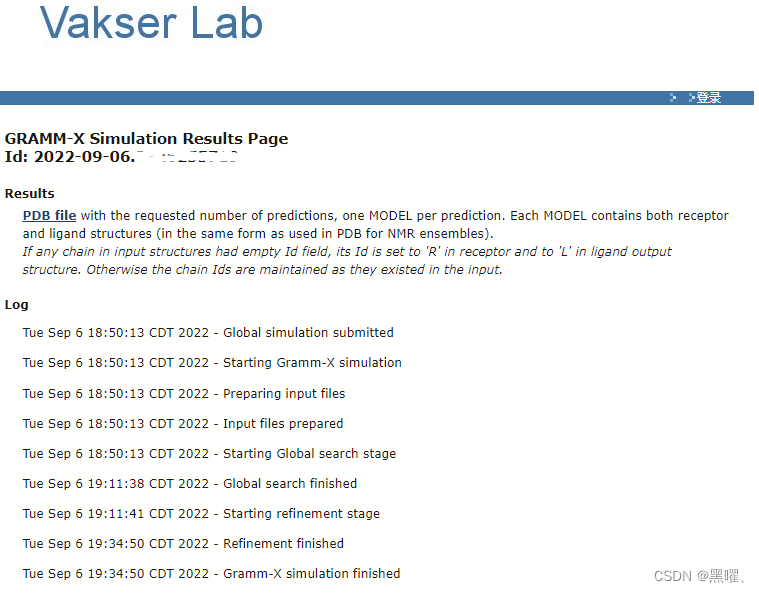
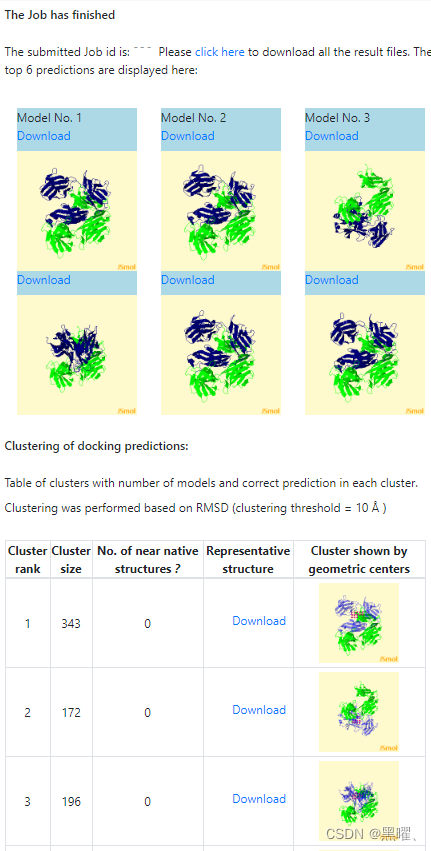
新版GRAMM结果页面就有结构展示更人性化,前面展示排名前6位的预测;
后面是每个簇中的模型数量和正确预测的簇表,根据均方根值(聚类阈值=10Å)执行聚类。
参考视频(虽然很方便,但GRAMM未必准):
https://www.bilibili.com/video/BV1Qq4y1o7cX?spm_id_from=333.337.search-card.all.click&vd_source=358bb9e0bb6e197db92319e1fb83b216
ZDOCK-分子对接
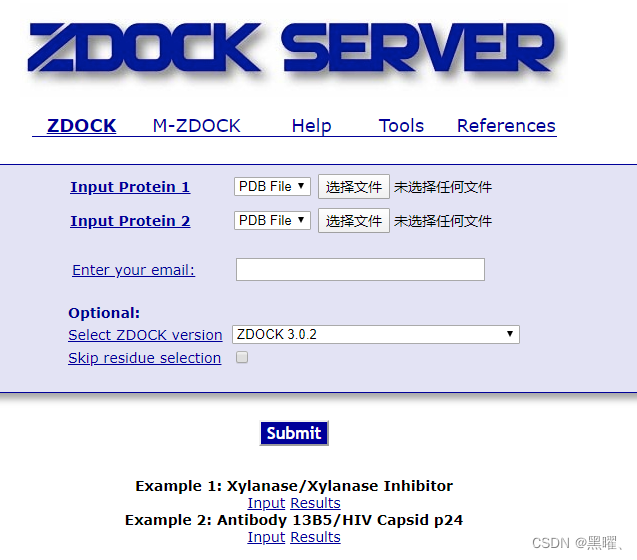
网页版:
https://zdock.umassmed.edu/
第二封邮件,点击链接进入下载界面。
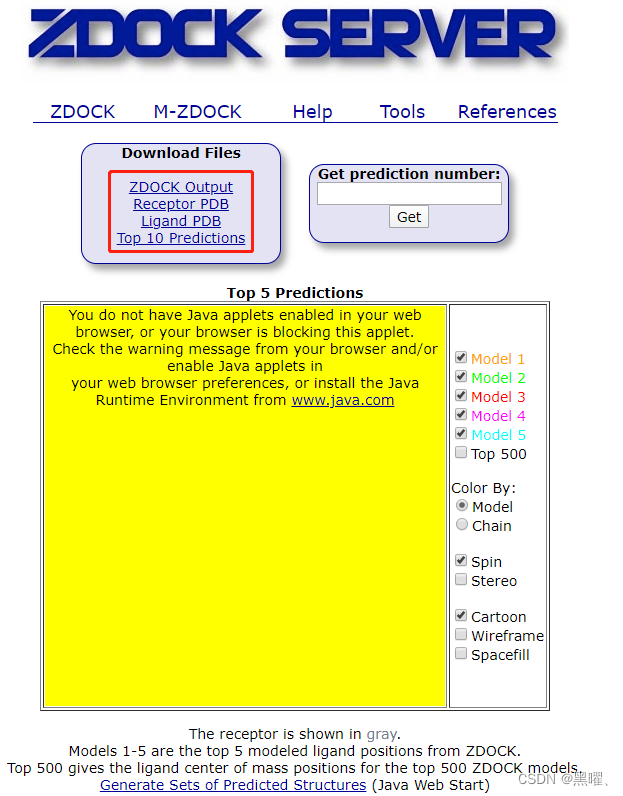
Download Files中有四个选项:
ZDOCK Output:包含有配置以及打分信息,从第6行开始,最后一列为打分,红框标出
Receptor PDB:输入的受体蛋白文件
Ligand PDB:输入的配体蛋白文件
Top 10 Predictions:打分最高的10个Pose
点击Top 10 Prediction下载结果,解压文件,使用可视化软件进行查看,Pymol,Chimera,VMD等等。
软件:
https://zlab.umassmed.edu/zdock/
下载的对接结果PDB文件可以用可视化的软件打开查看如PyMOL、Chimera、Discovery Studio、Schrodinger等,或者用下面提到的在线工具PDBePISA。
PDBePISA - 对接结果分析
https://www.ebi.ac.uk/msd-srv/prot_int/

上传成功后,点击“Interfaces”查看相互作用面情况。
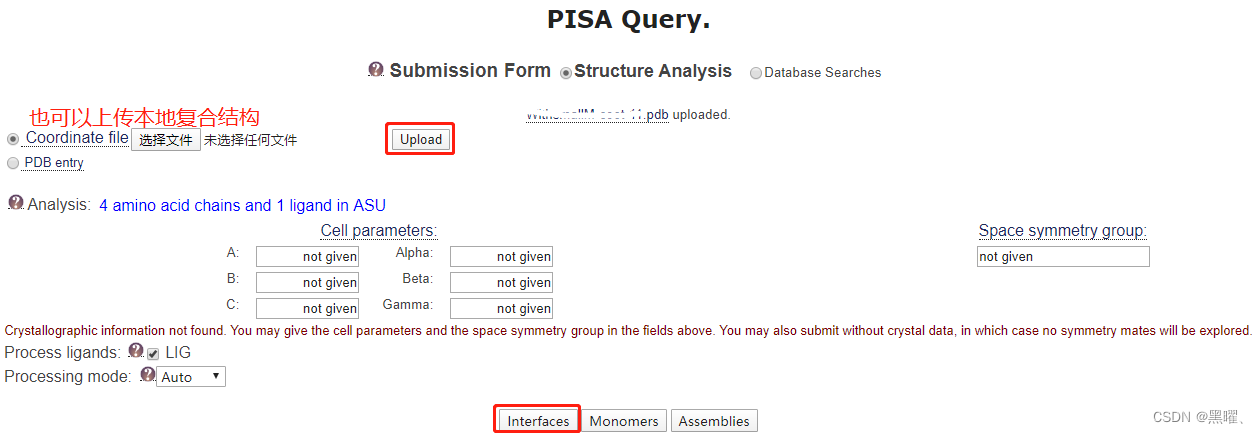
结合面列表(Interface List)里可以查看蛋白质相互作用面的面积大小(interface area, Å2),以及该种对接方式下自由能的高低(ΔiG kcal/mol),自由能越低说明结构越稳定。通常小于零的自由能才对应一个有意义的对接结果。点击“Download”可以下载如查看具体结合位点,点击“Details”查看更多信息。点击“view”可以看对接的图片。
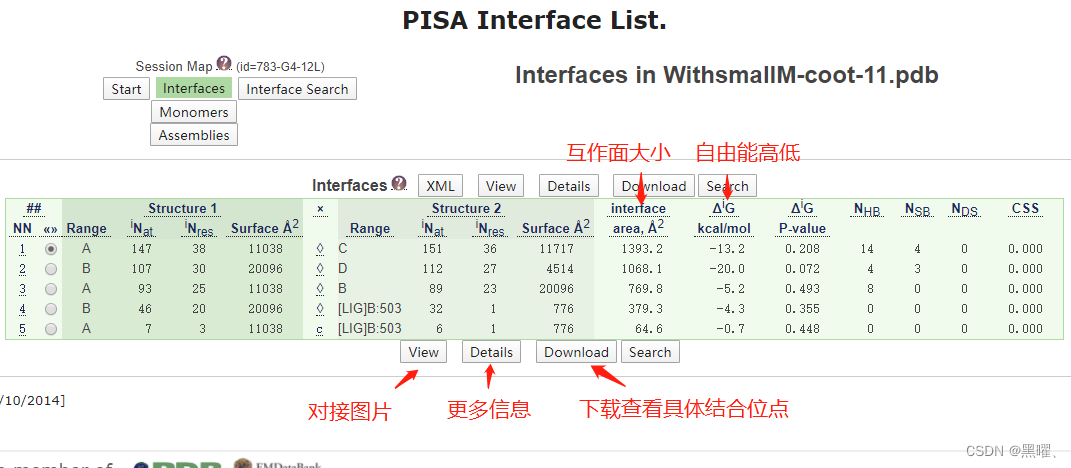
点击Details:
①“Interface Summary ”给出了对接总体的原子数、残基数、溶剂化自由能的数据及比例。
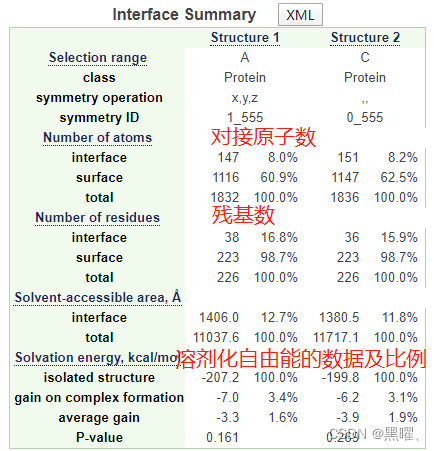
②“Hydrogen bonds”可以查看相互作用面上形成的所有氢键,以及形成氢键的原子和化学键的键长。
③“Interfacing residues”给出了两个蛋白质中所有参与相互作用的残基、残基参与的化学键(如氢键H、盐键S、二硫键D、共价键C)、残基埋入相互作用面的面积比,竖线越多说明埋入面积占比越大。
三种数据均可以下载XML文件。
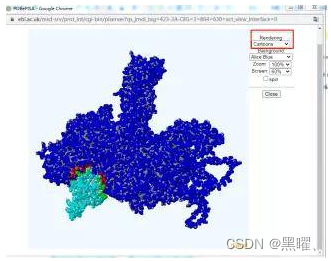
点击View页面还提供了Jmol插件,以图像化显示相互作用面和作用面上的残基。
Rending可以变换不同的显示形式。
参考:
http://news.sohu.com/a/511904965_120195181

旨在为数千万中国开发者提供一个无缝且高效的云端环境,以支持学习、使用和贡献开源项目。
更多推荐



 4
4 1
1- 0



 已为社区贡献2条内容
已为社区贡献2条内容

所有评论(0)